Pharmacology: Pharmacodynamics: Pharmacodynamic effects tested in an iron balance metabolic study with deferasirox tablets for oral suspension formulation of 10, 20 and 40 mg/kg/day was able to induce net iron excretion (0.119, 0.329 and 0.445 mg Fe/kg body weight/d, respectively) within the clinically relevant range (0.1 to 0.5 mg Fe/kg/day). Iron excretion was predominantly fecal.
Daily treatment with deferasirox (tablets for oral suspension) at doses of 20 and 30 mg/kg for one year in frequently transfused adult and pediatric patients with beta-thalassemia led to reductions in indicators of total body iron; liver iron concentration was reduced by about 0.4 and 8.9 mg Fe/g liver (biopsy dry weight) on average, respectively, and serum ferritin was reduced by about 36 and 926 μg/L on average, respectively. At these same doses the ratios of iron excretion: iron intake were 1.02 (indicating net iron balance) and 1.67 (indicating net iron removal), respectively. Deferasirox (tablets for oral suspension) induced similar responses in iron-overloaded patients with other anemias. Daily doses of 10 mg/kg for one year could maintain liver iron and serum ferritin levels and induce net iron balance in patients receiving infrequent transfusions or exchange transfusions.
The effect of 20 and 40 mg/kg of deferasirox (tablets for oral suspension) on QT interval was evaluated in a single-dose, double-blind, randomized, placebo-and active-controlled (moxifloxacin 400 mg), parallel group study in 182 healthy male and female volunteers aged 18 to 65 years. No evidence of prolongation of the QTc interval was observed in this study; however, the relevance of this study to long-term deferasirox use is unknown.
In patients with non-transfusion-dependent thalassemia syndromes and iron overload, treatment with deferasirox (tablets for oral suspension) at a dose of 10 mg/kg/day for one year led to a reduction in mean liver iron concentration from baseline by -3.80 mg Fe/g dw, while an increase of 0.38 mg Fe/g dw was observed in patients treated with placebo. In addition, treatment with deferasirox at a dose of 10 mg/kg/day for one year led to a reduction in mean serum ferritin from baseline by -222.0 microgram/L, while an increase of 114.5 microgram/L was observed in patients treated with placebo.
In patients with cardiac iron deposition (MRI T2* <20 ms), treatment with deferasirox (tablets for oral suspension) was shown to remove cardiac iron as demonstrated by progressive improvements in T2* values over 3 years of observation. In patients without cardiac deposition, deferasirox was shown to prevent clinically relevant cardiac iron deposition (maintenance of T2* at >20 ms) up to 1 year of observation, despite significant ongoing transfusion exposure.
In vitro: Affinity and selectivity of deferasirox for iron were assessed by potentiometric measurements, spectrophotometric titrations and cyclic voltammetry. Deferasirox has a high affinity for iron(III) with an overall affinity constant for the 1:2 complex (one Fe atom and two deferasirox molecules) in aqueous solution of 36.9 (logβ
2). Conversely, the affinity for iron(II) with a logβ
2 of 14.0 is low.
In a cell culture system using iron-loaded rat heart myocytes, deferasirox and deferoxamine showed similar potencies to remove iron at concentrations up to 80 μmol/L, which is a concentration that was achieved in human plasma following administration of efficacious doses.
In vivo: The potent and specific ability of deferasirox to mobilize tissue iron and to promote its excretion has been demonstrated in several animal studies. In the non iron-loaded, bile duct-cannulated rat, single oral doses of 25, 50 and 100 mg/kg deferasirox tablets for oral suspension showed a rapid response within the first three hours after administration of the compound. A protracted action of biliary iron excretion was noted, extending beyond 24 hours for the high doses of 50 and 100 mg/kg. Furthermore, iron excretion was dose dependent. The efficiency of iron excretion, defined as the amount of iron excreted as a percentage of the theoretical iron binding capacity of the dose, was higher than previously tested compounds (deferoxamine s.c. 2-4% and oral L1 2%), and amounted to 18.3% for the 25 mg/kg dose, which showed the highest effect.
In iron-overloaded marmosets receiving 14, 28, 56 or 112 mg/kg deferasirox, significantly higher fecal iron was measured for the doses of 56 and 112 mg/kg even two days after administration of deferasirox, corroborating the prolonged action found in rats. In addition, a dose dependent increase of iron excretion and superior efficacy of deferasirox compared to other chelators was found. With both animal models the bulk of the iron was excreted into bile (rat) or feces (marmoset) with less than 15% of the total iron found in urine, indicating that the iron complex is mainly cleared by the liver.
Radioactive iron given intravenously as deferasirox-iron complex was excreted in feces By inference, this suggests that iron complexes formed with deferasirox in the blood are also cleared by the liver.
Chronic administration of deferasirox to rats and marmosets demonstrated effective removal of iron from the liver, the main storage organ for iron. Conversely, in marmosets, deferasirox did not reduce liver zinc or liver copper levels. Likewise, zinc and copper levels in kidney were not found to be negatively affected, whereas kidney iron levels were reduced by approximately 40% in males and 30% in females at the highest dose of 80 mg/kg tested.
Safety Pharmacology: In the course of its safety evaluation, it could be shown in rat that deferasirox does not promote the uptake of dietary iron. A wide range of safety pharmacology studies has been conducted to assess the effects of deferasirox on behavior, cardiovascular, renal, and respiratory systems.
In mice, deferasirox effects on CNS function included ataxia, slight head tremors, and effects on step-through passive avoidance.
In vitro receptor-binding assays showed that deferasirox at 10 μmol/L only interacted weakly with kainate receptors and the channel site of NMDA receptors.
Renal evaluations in the rat after single doses up to 1000 mg/kg revealed no effects on the excretion of Cl
-, Na+ and K+ and urine volume. Intraduodenal administration of deferasirox at doses up to 1000 mg/kg to anesthetized rats demonstrated no effect on respiratory rate, tidal volume or minute volume. A variety of
in vitro and
in vivo studies were conducted to explore possible cardiovascular effects of deferasirox.
The data from the
in vitro studies with isolated atria, heart or Purkinje fibers demonstrated no consistent pattern of changes. In an
in vivo dog telemetry study, deferasirox demonstrated an increase in mean heart rate only at an exposure (Cmax) of 734 μmol/L. No ECG changes were observed in marmoset toxicity studies after 4 weeks (130 mg/kg; Cmax of 127-135 μmol/L) or 39 weeks (80 mg/kg; Cmax of 64-81 μmol/L). Neither the hERG assay nor the dog study showed any evidence for QTc prolongation potential.
Mechanism of Action: JADENU (deferasirox) is an orally active chelator that is highly selective for iron (as Fe
3+). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Although its highest affinity is for iron, deferasirox has a significant affinity for aluminum. Deferasirox has very low affinity for zinc and copper, and there are variable decreases in the serum concentration of these trace metals after the administration of deferasirox. The clinical significance of these decreases is uncertain.
JADENU (deferasirox) is an orally active iron chelating agent. The core structure of deferasirox is an N-substituted bis-hydroxyphenyl-triazole, representative of a new class of tridentate and iron selective chelators. In this structure, potent iron-coordinating atoms are arranged in a geometry optimal for the formation of tridentate complexes.
Clinical Trials: The following information is based on clinical trials conducted with deferasirox tablets for oral suspension. JADENU contains the same active ingredient as EXJADE dispersible tablets for oral suspension; however, the exposure is 30% higher in the JADENU tablets. Currently, there are no clinical trial data in patients administered JADENU tablets; however, JADENU has been evaluated in healthy volunteer trials.
Comparative Bioavailability Studies: The bioavailability of deferasirox tablets was compared to that of EXJADE dispersible tablets for oral solution in a randomized, two-way, comparative bioavailability study in 32 healthy adult male and female subjects. Single doses of deferasirox were administered as 1080 mg (3 x 360
mg) tablets swallowed whole with water, or as 1500 mg (3 x 500 mg) EXJADE tablets, which were dispersed in a glass of water. The results of the study demonstrated similar values for AUC
T for the two dosage forms; however, C
max was 30% higher for the tablets as compared to EXJADE dispersible tablets for oral solution. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Study demographics and trial design: Study 0107, was a 1-year, multi-centre, open-label, randomized, Phase III, active comparator control study to compare deferasirox tablets for oral suspension and deferoxamine in patients with β-thalassemia and transfusional hemosiderosis. Patients ≥2 years of age were randomized in a 1:1 ratio to receive either oral deferasirox tablets for oral suspension at starting doses of 5, 10, 20 or 30 mg/kg once daily or subcutaneous DESFERAL*
1(deferoxamine) at starting doses of 20 to 60 mg/kg for at least 5 days per week based on LIC (liver iron concentration) at baseline (2 to 3, >3 to 7, >7 to 14 and >14 mg Fe/g dry weight (dw)). Patients randomized to deferoxamine who had LIC values <7 mg Fe/g dw were permitted to continue on their prior deferoxamine dose, even though the dose may have been higher than specified in the protocol. Consequently, the ratio of deferasirox tablets for oral suspension to deferoxamine doses for the two lower LIC categories was disproportionately low (1:4) compared to the two upper LIC categories (1:2). A total of 586 patients were randomized and treated (including 154 patients <16 years of age and received either deferasirox tablets for oral suspension (296 patients) or deferoxamine (290 patients). There were no major differences in the baseline demographic characteristics between the groups. In both groups more than 97% of patients had received prior chelation therapy.
Approximately two-thirds of each group was heavily iron overloaded as evidenced by an LIC value > 7 mg Fe/g dw at baseline.
Study 0108 was an open-label, non-comparative, phase II trial of efficacy and safety of deferasirox tablets for oral suspension given for 1 year to patients with chronic anemias and transfusional hemosiderosis unable to be treated with deferoxamine. Similar to Study 0107, patients received 5, 10, 20, or 30 mg/kg per day of deferasirox tablets for oral suspension based on baseline LIC. A total of 184 patients (adult and pediatric) were treated in this study: 85 patients with β-thalassemia and 99 patients with other congenital or acquired anemias (myelodysplastic syndromes, n=47; Diamond-Blackfan syndrome, n=30; other, n=22). Nineteen percent (N=35) of patients were <16 years of age (11 patients were ≥ 2 - < 6 years, 11 patients were 6 - < 12 years, and 13 patients were 12 - < 16 years) and 16% (N=30) of patients were ≥ 65 years of age. Thirty-seven patients had not received prior chelation therapy.
Study 0109 was a 1-year, open-label, randomized, Phase II, active comparator control study to compare deferasirox tablets for oral suspension and deferoxamine in patients with sickle cell disease and transfusional hemosiderosis. As in Study 0107, patients received 5, 10, 20, or 30, mg/kg per day of deferasirox tablets for oral suspension or subcutaneous deferoxamine at doses of 20 to 60 mg/kg for 5 days per week based on baseline LIC. The primary objective of this study was safety and tolerability of deferasirox in this patient population. The population examined in study 0109 was adult and pediatric patients with sickle cell disease and chronic iron overload from repeated blood transfusions. This population included individuals receiving intermittent or regular transfusions. A total of 195 patients were randomized to receive either deferasirox tablets for oral suspension (132 patients) or deferoxamine (63 patients) with the following distribution by age group: 7 patients were 2-< 6 years; 45 patients were 6 - < 12 years; 46 patients were 12 - <16 years; 96 patients were ≥ 16 years. There were no major differences in the patient populations randomized to receive either deferasirox or deferoxamine with regard to baseline demographics and disease characteristics. In both groups about 60% of patients had received prior chelation therapy. A somewhat higher percentage of deferasirox patients were heavily iron overloaded (LIC value > 7 mg Fe/g dw) at baseline when compared with deferoxamine (deferasirox 64%; deferoxamine 49%).
Relevant demographic characteristics for these studies are shown in Table 2 and Table 3. (See Table 2 and Table 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Study results: In the primary efficacy study 0107, treatment duration was 12 months. LIC, an accepted indicator of total body iron burden, was assessed at baseline and after 12 months of therapy by liver biopsy or non-invasively by biomagnetic susceptometry. Success rate, the primary efficacy endpoint, was defined as a reduction in LIC of ≥ 3 mg Fe/g dw for baseline values ≥ 10 mg Fe/g dw, reduction of baseline values between 7 and < 10 to < 7 mg Fe/g dw, or maintenance or reduction for baseline values <7 mg Fe/g dw. Deferasirox was to be declared non-inferior to deferoxamine if the lower limit of the 95% confidence interval (two-sided) of the difference in success rates was above -15%. (See Table 4.)
Click on icon to see table/diagram/image
The primary efficacy population consisted of 553 patients (deferasirox n=276; deferoxamine n=277) who had LIC evaluated at baseline and 12 months or discontinued due to an AE. Of these 553 patients, 56 patients were < 6 years; 130 patients were 6 - < 12 years; 106 patients were 12 - <16 years; 261 patients were ≥ 16 years and <65 years. The overall success rates were 52.9% for deferasirox and 66.4% for deferoxamine with a difference of -13.5 in success rates and a 95% CI of [-21.6, -5.4]. Non-inferiority to deferoxamine was not achieved because the lower limit of the CI was below -15%. However, non-inferiority was demonstrated in a group of patients with baseline LIC levels ≥ 7 mg Fe/g dw who were allocated to the higher dose groups (deferasirox tablets for oral suspension doses of 20 or 30 mg/kg and deferoxamine doses of ≥ 35 mg/kg. The success rates with deferasirox and deferoxamine were 58.6% and 58.9%, respectively, and the lower limit of the 95% CI (-10.2%) was above the non-inferiority threshold of -15% (see Table 4).
In patients with LIC ≥ 7 mg Fe/g dw who were treated with deferasirox tablets for oral suspension 20 to 30 mg/kg per day a statistically significant reduction in LIC from baseline was observed (-5.3 ± 8.0 mg Fe/g dw, p<0.001, t-test) which was not statistically significantly different from deferoxamine (-4.3 ± 5.8 mg Fe/g dw, p = 0.367). (See Table 5.)
Click on icon to see table/diagram/image
Reduction of LIC and serum ferritin were observed with deferasirox tablets for oral suspension doses of 20 to 30 mg/kg. Deferasirox tablets for oral suspension doses below 20 mg/kg/day failed to provide consistent lowering of LIC and serum ferritin levels (Figure 1). Therefore, a starting dose of 20 mg/kg/day of deferasirox tablets for oral suspension is recommended (see Dosage & Administration). (See figure.)
Click on icon to see table/diagram/image
The results of the primary efficacy study are supported by the second major efficacy study, study 0108. The primary endpoint was to demonstrate a success rate significantly greater than 50% with deferasirox. In the total population, the success rate (50.5%) was not statistically significantly higher than 50%. However, in patients with LIC ≥ 7 mg Fe/g dw for whom both baseline and end of study LIC was available and who received deferasirox tablets for oral suspension 20 to 30 mg/kg per day, the success rate was 58.5% [p=0.022 (50.3, 66.6)] and there was a statistically significant reduction in the absolute LIC from baseline to end of study (-5.5 ±
7.4 mg Fe/g dw, p < 0.001, t-test). There was also a dose dependent effect on serum ferritin and the ratio of iron excretion to iron intake from doses of 5 to 30 mg/kg per day.
The primary objective of study 0109 was safety and tolerability (see Adverse Reactions).
A total of 132 patients were treated with deferasirox and 63 patients with deferoxamine. At the time of the 6-month interim analysis, dose-dependent increases in the ratio of iron excretion to iron intake from doses of 5 to 30 mg/kg per day of deferasirox tablets for oral suspension were observed. At the end of the study, the mean change in LIC in the per protocol-1 (PP-1) population, which consisted of patients who had at least one post-baseline LIC assessment, was -1.3 mg Fe/g dry weight for patients receiving deferasirox (n=113) and -0.7 mg Fe/g dry weight for patients receiving deferoxamine (n=54).
In an analysis of 192 beta-thalassemia patients dose escalated up to a maximum dose of 40 mg/kg/day of deferasirox tablets for oral suspension (treated for up to 32 weeks), a further decrease in serum ferritin of 11.9% was observed (from the start of dosing >30 mg/kg/day). This was based on a pooled analysis of patients who were exposed to doses greater than 30 mg/kg/day of deferasirox tablets for oral suspension in the key registration trials and their ongoing long-term extensions (Studies 0107/E, 0108/E, and 0109/E), and in another large clinical trial and its ongoing long-term extension (2402/E).
A cardiac sub-study was conducted as part of a Phase IV study. The cardiac sub-study was a one year, prospective, open-label, single-arm study which included two cohorts of severely iron overloaded β-thalassemia patients with LVEF values ≥56%: 114 patients with baseline T2* values >5 to <20 ms indicating myocardial siderosis (treatment cohort) and 78 patients with myocardial T2* ≥20 ms indicating no clinically significant cardiac iron deposition (prevention cohort). In the treatment cohort, the deferasirox tablets for oral suspension starting dose was 30 mg/kg/day, with escalation to a maximum of 40 mg/kg/day. In the prevention cohort, the deferasirox tablets for oral suspension starting dose was 20-30 mg/kg/day, with escalation to a maximum of 40 mg/kg/day. The primary endpoint of the cardiac sub-study was the change in T2* at one year. In the treatment cohort, T2* (geometric mean ± coefficient of variation) significantly increased from a baseline value of 11.2 ms ± 40.5% to 12.9 ms ± 49.5%, representing a significant improvement of 16% (p <0.0001). In the treatment cohort, improvement in T2* was observed in 69.5% of patients and stabilization of T2* in 14.3% of patients. LVEF remained stable and within the normal range: 67.4 ± 5.7% to 67.1 ± 6.0%. In the prevention cohort, myocardial T2* remained within the normal range and was unchanged from a baseline value of 32.0 ms ± 25.6% to 32.5 ms ± 25.1% (+2%; p = 0.565) indicating that daily treatment with deferasirox can prevent cardiac iron loading in β-thalassemia patients with a history of high transfusion exposure, and regular, ongoing transfusions.
Patients in the treatment cohort of the 1-year core study had the option to participate in two 1-year extensions. Over a three-year treatment duration period, there was a statistically significant (p<0.0001), progressive improvement in the geometric mean of cardiac T2* from baseline overall, in the severe cardiac iron overload sub-group, which is associated with a high risk of cardiac failure (T2* >5 to <10 ms), and in the mild to moderate cardiac iron overload sub-group (T2* 10 to <20 ms) (Table 6). Using the geometric mean ratio, the T2* increase was 43% above baseline in all patients, 37% increase from baseline in the T2* >5 to <10 ms sub-group, and 46% increase from baseline in the T2* 10 to <20 ms sub-group. Continuous treatment with deferasirox tablets for oral suspension for up to 3 years at doses >30 mg/kg/day effectively reduced cardiac iron in thalassemia major patients with myocardial siderosis as shown by the number of patients who normalized their T2* or improved to a category associated with a lower risk of cardiac failure (Table 7). (See Table 6 and Table 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A randomized, double-blind, placebo-controlled study to compare deferasirox tablets for oral suspension and placebo was conducted in patients with non-transfusion-dependent thalassemia syndromes and iron overload. Patients ≥10 years of age were enrolled in the study in a 2:1:2:1 randomization to receive either deferasirox tablets for oral suspension 5 mg/kg/day or deferasirox 10 mg/kg/day or matching placebo.
Transfusion independency of the patients was confirmed by the fact that blood transfusions 6 months prior to study start were not allowed and patients were excluded if a regular transfusion program was anticipated during the study. Iron overload was diagnosed by a serum ferritin >300 microgram/L at screening (two consecutive values at least 14 days apart from each other) and LIC ≥5 mg Fe/g dw measured by R2 MRI at screening. All patients with non-transfusion-dependent thalassemia syndromes were allowed with the exception of patients with HbS-variants or those whose clinical condition allowed phlebotomy.
In total, 166 patients were randomized. Demographics were well balanced. The main underlying disease was beta-thalassemia intermedia in 95 (57.2%) patients and HbE beta-thalassemia in 49 (29.5%) patients. The primary efficacy endpoint of change in liver iron concentration (LIC) from baseline to Week 52 was statistically significant in favor of both deferasirox treatment groups compared with placebo (Table 8). Furthermore, a statistically significant dose effect of deferasirox tablets for oral suspension was observed in favor of the 10 mg/kg/day dose. (See Table 8.)
Click on icon to see table/diagram/image
The primary efficacy result was supported by additional analyses which showed a clear dose-response effect; this was reflected by a greater percentage of patients with an LIC decrease of ≥3 mg Fe/g dw in the 10 mg/kg/day deferasirox tablets for oral suspension group compared to the 5 mg/kg/day deferasirox group (56.4% versus 32.7%, respectively). In addition, a reduction of ≥30% in LIC between baseline and Week 52 was reported in approximately twice as many patients in the 10 mg/kg/day deferasirox tablets for oral suspension group (49.15%) compared to the 5 mg/kg/day deferasirox tablets for oral suspension group (25.5%). After one year of treatment, 27.3% of patients in the 10 mg/kg/day deferasirox tablets for oral suspension group and 14.5% of patients in the 5 mg/kg/day deferasirox tablets for oral suspension group achieved an LIC of < 5mg Fe/g dw.
In the deferasirox treated groups, three pregnancies were reported among 45 female patients of child-bearing potential; one of these occurred despite concomitant oral contraceptive use. deferasirox may decrease the efficacy of hormonal contraceptives (see Interactions).
Pharmacokinetics: JADENU tablets are a strength-adjusted formulation of deferasirox with higher bioavailability compared to the tablet for oral suspension formulation. After strength-adjustment, the JADENU tablet formulation (i.e., 360 mg strength) had comparable bioavailability to the tablet for oral suspension formulation (i.e., 500 mg strength) with respect to the mean area under the plasma concentration time curve (AUC) under fasting conditions. The Cmax was increased by 30% (90% CI 20.3% to 40.0%), however a clinical exposure/response analysis revealed no effects of clinical relevance.
Absorption: Based on studies in patients with the tablet for oral suspension, deferasirox is absorbed following oral administration with a median time to maximum plasma concentration (t
max) of about 1.5 to 4 hours. In healthy volunteers, the JADENU tablet formulation showed comparable t
max. The C
max and AUC of deferasirox increase approximately linearly with dose after both single administration and under steady-state conditions. Exposure to deferasirox increased by an accumulation factor of 1.3 to 2.3 after multiple doses with the tablet for oral suspension formulation.
The absolute bioavailability (AUC) of deferasirox tablets for oral suspension is 70% compared to an intravenous dose. Bioavailability of deferasirox in JADENU tablets was 36% greater than with EXJADE dispersible tablets for oral suspension.
A food-effect study involving administration of the JADENU tablets to healthy volunteers under fasting conditions and with a light (i.e., a whole wheat English muffin with jelly and a glass of skim milk) or high-fat (fat content >50% of calories) meal indicated that the AUC and C
max were slightly decreased after a light meal (by 11% and 16%, respectively). After a high-fat meal, AUC and C
max were increased by 18% and 29%, respectively. The increases in C
max due to the change in formulation and due to the effect of a high-fat meal may be additive and therefore, it is recommended that JADENU should be taken on an empty stomach or with a light meal (see Dosage & Administration).
Distribution: Deferasirox is highly (~99%) protein bound almost exclusively to serum albumin. The percentage of deferasirox confined to the blood cells was 5% in humans. The volume of distribution at steady state (Vss) of deferasirox is 14.37 ± 2.69 L in adults.
Metabolism: Glucuronidation is the main metabolic pathway for deferasirox, with subsequent biliary excretion. Deconjugation of glucuronidates in the intestine and subsequent reabsorption (enterohepatic recycling) is likely to occur. Deferasirox is mainly glucuronidated by UGT1A1 and to a lesser extent UGT1A3. CYP450-catalysed (oxidative) metabolism of deferasirox appears to be minor in humans (about 8%). No evidence for induction or inhibition of CYP450 enzymes (CYP1A1, CYP1A2 and CYP2D6) at therapeutic doses has been observed. No inhibition of deferasirox metabolism by hydroxyurea was observed in an
in vitro study. Deferasirox undergoes enterohepatic recycling.
Excretion: Deferasirox and metabolites are primarily (84% of the dose) excreted in the feces. Renal excretion of deferasirox and metabolites is minimal (8% of the dose). The mean elimination half-life (t
½) ranged from 8 to16 hours following oral administration.
Special Populations and Conditions: Hepatic Insufficiency: The average AUC of deferasirox in 6 subjects with mild hepatic impairment (Child-Pugh A) increased 16% over that found in 6 subjects with normal hepatic function, while the average AUC of deferasirox in 6 subjects with moderate hepatic impairment (Child-Pugh B) increased 76% over that found in 6 subjects with normal hepatic function. The average C
max of deferasirox in subjects with mild or moderate hepatic impairment increased 22% over that found in subjects with normal hepatic function (see Dosing Considerations under Dosage & Administration, Warnings and Precautions). Efficacy of deferasirox was not studied in this pharmacokinetic investigation of subjects with hepatic impairment.
Pharmacokinetics and disposition of
14C-labeled and non-radiolabeled deferasirox, its metabolites, and the respective iron complex Fe-[deferasirox]
2 were investigated comprehensively in mice, rats, dogs and marmosets, including in humans. The fate of deferasirox appears similar in all species including human, with minor differences.
The extent of oral absorption and bioavailability of deferasirox was investigated after intravenous and oral administration of
14C-labeled deferasirox in mice, rats and marmosets and with nonradiolabeled deferasirox in dogs (see Table 9).
Click on icon to see table/diagram/image
Using specific and sensitive analytical methods, deferasirox, metabolites and iron complex Fe-[deferasirox]
2 were quantified in various biological matrices. Orally administered deferasirox is well and rapidly absorbed in all species investigated including man. Oral bioavailability is substantial if not complete, with dose over-proportional increase in rodents and female rabbits, probably due to saturation of elimination processes. In marmosets and humans the systemic exposure to deferasirox increased proportionally to the dose. No unexpected accumulation and no significant gender differences were observed in the pharmacokinetics. Deferasirox is the major active circulating moiety in the animal species and human, and is considered to contribute most to the overall iron elimination
in vivo. Two hydroxy metabolites of deferasirox (M1 and M2), which were found to be able to form iron complexes
in vitro are considered to contribute negligibly to the overall iron excretion capacity of deferasirox.
Deferasirox in the blood was mainly confined to the plasma compartment of human and dog (≥ 90%), and to a lesser extent in the rabbit, marmoset, rat and mouse. For the deferasirox iron complex, almost no uptake to blood cells was observed. Deferasirox and its iron complex were extensively (98%-99%) bound to plasma proteins and primarily to human serum albumin for all species including human.
Deferasirox shows a distribution pattern typical for a compound with a low volume of distribution: deferasirox was distributed throughout the body, but was mainly present intravascularly. Substantial levels were found in organs of the gastrointestinal tract and excretory organs. Deferasirox and/or its metabolites passed the blood-brain barrier to a very low extent only. The placental barrier was passed to a very low extent only. Deferasirox was enriched in the milk building up a milk-to-plasma ratio of up to 20. Suckling juvenile rats were distinctly exposed to deferasirox. The tissue distribution pattern in juvenile animals was qualitatively similar to that in mother animals. No notable retention was observed in any tissue or organ of the albino and pigmented rat.
Metabolism of deferasirox includes mainly glucuronidation (animals and human), and to a lesser extent cytochrome P450-catalysed hydroxylation, in human mainly by CYP1A1, CYP1A2 and CYP2D6. Direct glucuronidation of deferasirox to the acyl-glucuronide (M3) occurred predominantly by UGT1A1 and UGT1A3. Drug-drug interactions by deferasirox based on UGT isoenzymes are in principle possible when a second co-administered drug is metabolized solely or mainly by UGT1A1 or UGT1A3. Any inhibition or induction of the cytochrome P450 enzymes by co-medications is not expected to significantly affect the pharmacokinetics of deferasirox. The potential for drug-drug interactions between deferasirox and comedications via cytochrome P450 enzymes, and
via hepatic anion transport appears low. Based on the available data on the pharmacokinetic and disposition of deferasirox in animals and man, deferasirox appears to have a very low potential for induction of drug metabolizing enzymes in the liver.
Elimination of deferasirox and metabolites is rapid and complete. The elimination of the iron complex of deferasirox could not be determined in bile and/or feces due to its inherent instability in these matrices. Key elimination processes are hepatic metabolism and hepatobiliary elimination. Biliary elimination could be studied in rats only, but the findings are assumed to apply to higher animal species and humans as well. Hepatobiliary elimination may occur to some extent by first pass. There is evidence for enterohepatic recirculation of deferasirox and it metabolites. Enterohepatic recirculation can be ascribed to hepatobiliary elimination and intestinal hydrolysis of glucuronides to deferasirox. Deferasirox, its metabolites and the iron complex are anions, and seem to be eliminated largely
via bile by hepatic canalicular anion transport (as shown in data from mrp2-deficient (TR-) rats). Active transporters expressed at the canalicular membranes of the hepatocytes e.g. MRP2, MXR (also called BCRP) may be involved in the elimination of deferasirox, its iron complex and its metabolites.
Toxicology: Acute Toxicity Studies: Single oral doses of deferasirox tablets for oral suspension at 1000 mg/kg in mice and ≥ 500 mg/kg in rats resulted in mortality/morbidity. Single intravenous doses of deferasirox in mice resulted in mortality at 150 mg/kg. No mortality was observed in rats at the highest intravenous dose tested, 75 mg/kg.
Subacute Toxicity Studies: Mortality was observed at doses ≥ 200 mg/kg and at 100 mg/kg in the 2-week and 4-week rat study, respectively. Decreased tissue iron and changes in hematological parameters characteristic of a potent iron chelator were evident. Histopathologic findings of renal cortical tubular cytoplasmic vacuolation and gastrointestinal tract were common to both studies. Decreased hematopoiesis in the spleen, and splenic lymphoid depletion was observed after two weeks of administration. All effects were reversible following a nondosing phase. In a rat exploratory studies in which rats were iron overloaded or received diet supplemented with iron or findings were limited to pharmacological effects on tissue/serum iron levels.
In 2 and 4-week studies in marmosets, decreased tissue iron levels was observed at all doses of deferasirox. Effects on hematopoiesis were evident at 400 mg/kg after 2-weeks of administration and at 130 mg/kg after 4-weeks of treatment, vacuolar degeneration of the renal cortical tubules at doses ≥ 200 mg/kg and at 130 mg/kg in the 2-week and 4-week study, respectively. Vacuolation of intrahepatic bile duct cells and marked inflammation of gall bladder epithelium with fibrosis of the gall bladder wall and vacuolar hyperplasia of the epithelium was noted in a single animal at 130 mg/kg after 4 weeks treatment. All effects were reversible following a nondosing phase. In a two week exploratory study in marmosets preloaded with iron, no deferasirox related effects were observed. Dietary iron supplementation of marmosets did not reduce deferasirox effects.
Long Term Toxicity Studies: In a 26-week oral study in rats (with dietary iron supplementation) at doses of 0, 30, 80 or 180 mg/kg, mortality was observed at 180 mg/kg. Cataracts, characterized by lenticular degeneration and fragmentation, vacuole formation and/or lenticular epithelial hyperplasia were present at doses ≥ 80 mg/kg. Early lenticular changes were observed at 30 mg/kg. Cytoplasmic vacuolation of renal cortical tubular epithelium and splenic hematopoiesis occurred at 180 mg/kg. Ulceration/erosion of the glandular stomach was observed at ≥ 80 mg/kg. With the exception of the lenticular cataracts, all effects were reversible following a nondosing phase.
Oral administration of deferasirox to marmosets for 39 weeks at doses of 0, 20, 40 or 80 mg/kg resulted in mortality at 80 mg/kg. Histopathology findings at 80 mg/kg consisted of vacuolation of the hepatic bile duct cells; vacuolation and/or degeneration of the renal cortical tubules and dilatation of medullary tubules.
Fertility: Deferasirox at oral doses up to 75 mg/kg/day (which resulted in a drug exposure (plasma AUC) that was less than the maximum human value) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
Reproduction and Teratology: Deferasirox was not teratogenic in rats or rabbits treated with doses up to and exceeding the maximum tolerated doses. Increased skeletal variations were seen in rats at a maternotoxic dose of 100 mg/kg/day, which achieved a drug exposure (plasma AUC) that was similar to the maximum human value. No adverse effect on fetal development was observed in rabbits at a maternotoxic dose of 50 mg/kg/day, which achieved a drug exposure about 30% of the maximum human value.
In a rat study designed to evaluate for effects on pre- and post-natal development, rats were treated at doses up to 90 mg/kg/day, a dose lethal to maternal animals, from early gestation to end of lactation. This treatment resulted in an increase in the number of stillborn pups and reduced pup birth weight.
Mutagenicity: Deferasirox was negative in the Ames test and an
in vitro chromosome aberration assay with human peripheral blood lymphocytes. Positive responses were observed in an
in vitro (V79) micronucleus screening test and in a rat
in vivo bone marrow micronucleus assay, which may have been a result of altered hematopoiesis due to iron chelation. No response was observed in another rat
in vivo micronucleus assay (liver) with doses up to 250 mg/kg.
Carcinogenicity: Deferasirox was not carcinogenic in a 104-week study in Wistar rats or in a 26-week study in transgenic p53+/- heterozygous mice that were maintained on an iron-supplemented diet.
In the rat carcinogenicity study, rats were administered deferasirox daily for 2 years at doses up to 60 mg/kg resulting in plasma exposure that were 28 to 39% of human exposure at 20 mg/kg based on plasma AUC
0-24hr.
In the mouse oral carcinogenicity study, transgenic p53+/- heterozygous mice were treated daily for 26 weeks at doses up to 200 mg/kg in males and 300 mg/kg in females, which resulted in plasma exposures that were 122% and 210% of human exposure at 20 mg/kg, respectively, based on plasma AUC
0-24hr.
104-week rat carcinogenicity study: No deferasirox-related neoplastic or non-neoplastic lesions were detected.
26-week transgenic mouse carcinogenicity study: No deferasirox-related neoplastic lesions were observed. Non-neoplastic lesions observed in mice were generally similar to those observed in 26 week toxicity study in rats and included biliary hyperplasia and hepatic periportal inflammation.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image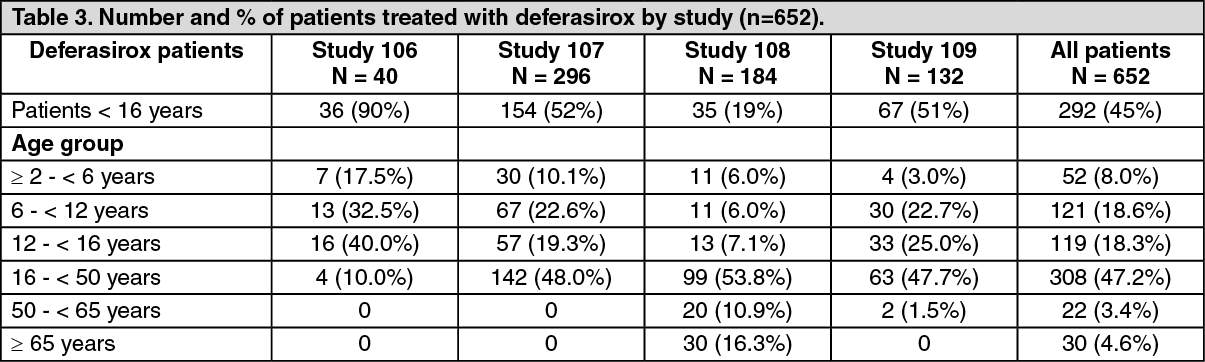 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image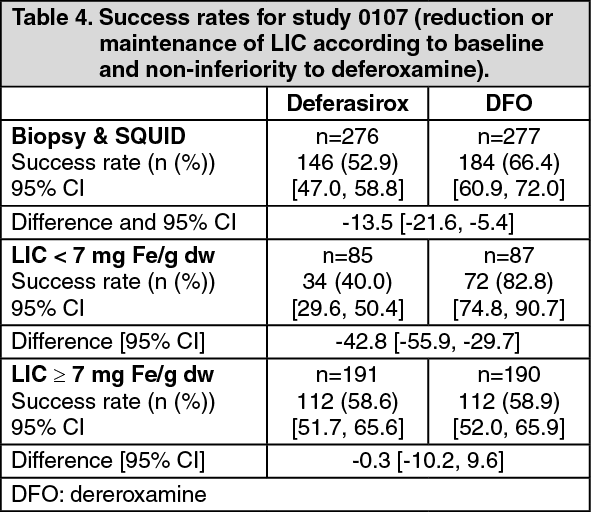 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image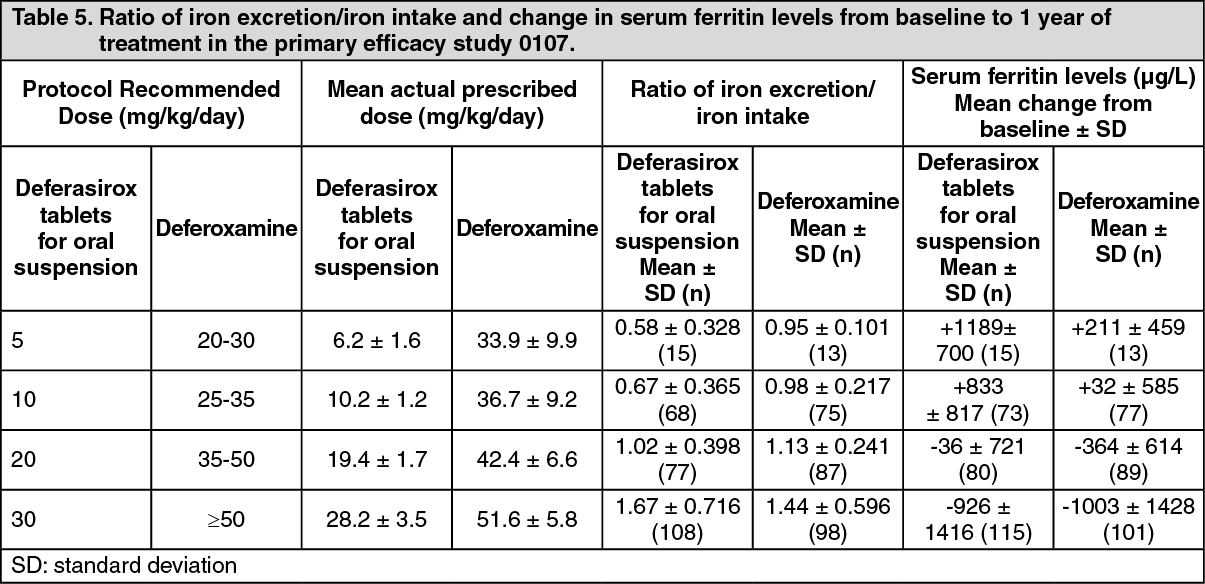 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image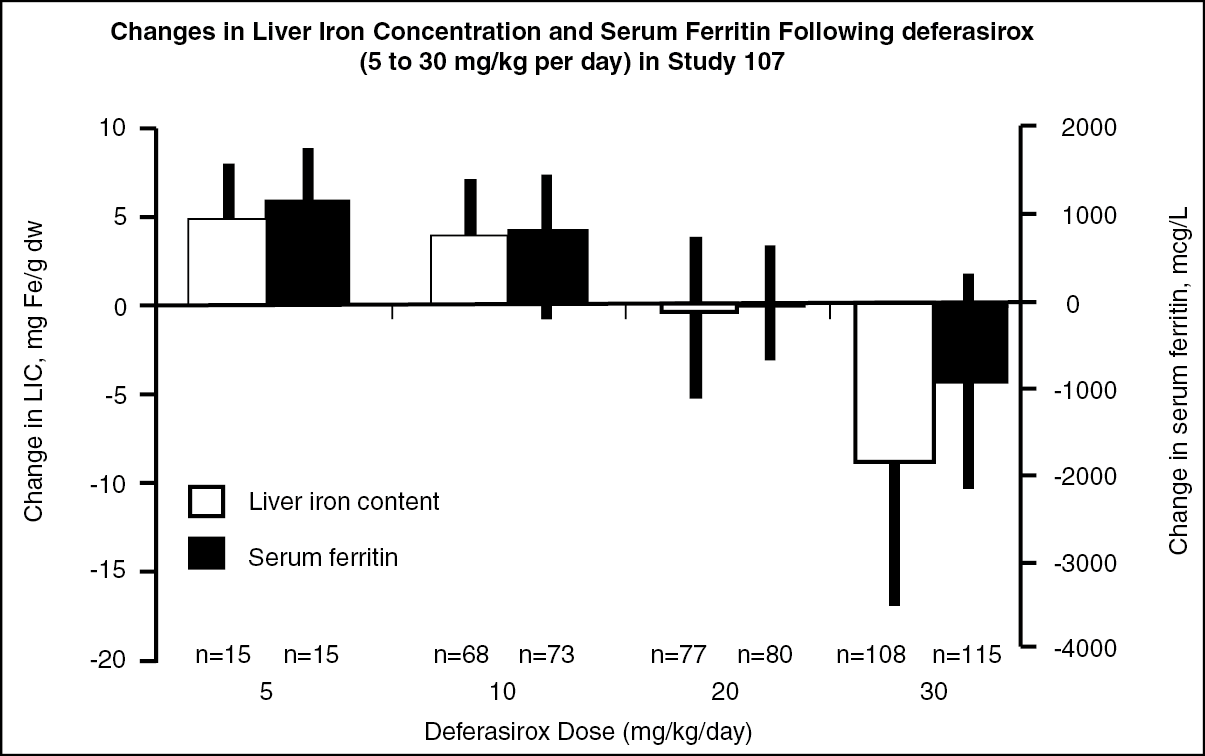 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image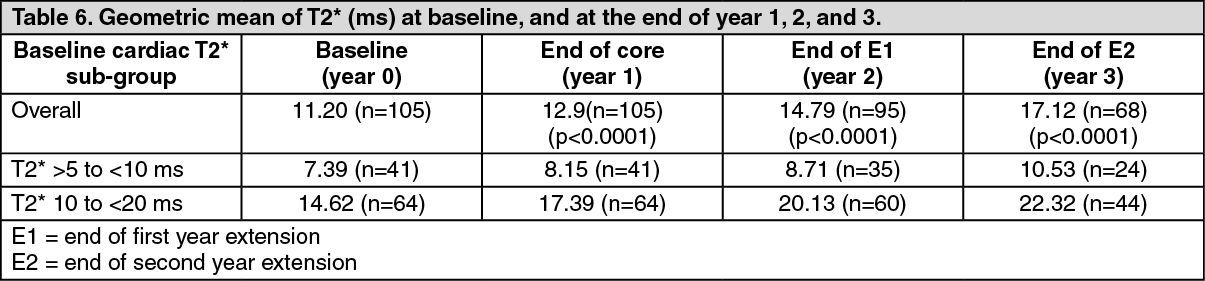 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image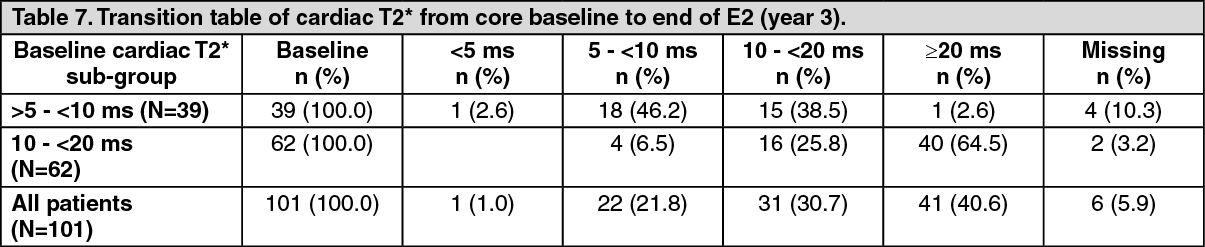 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image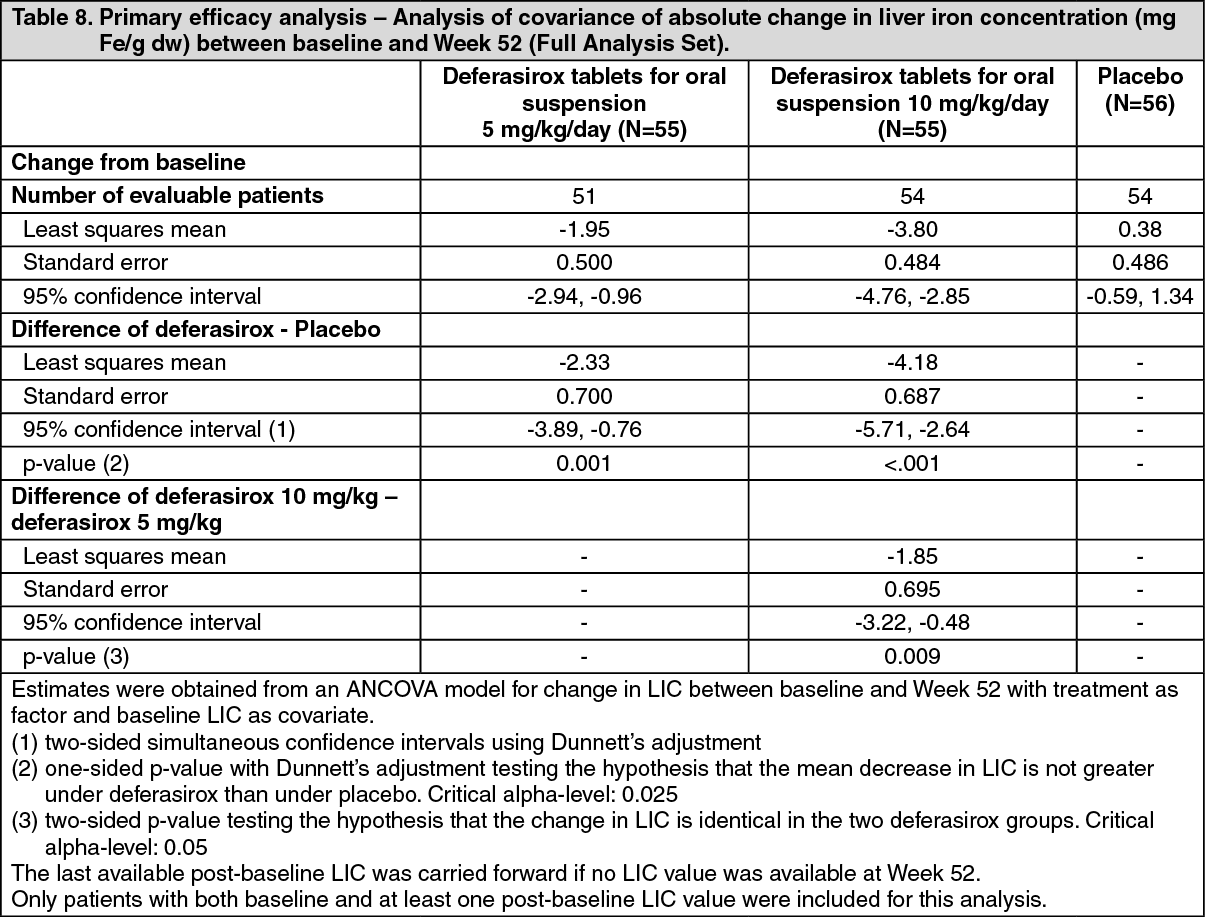 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image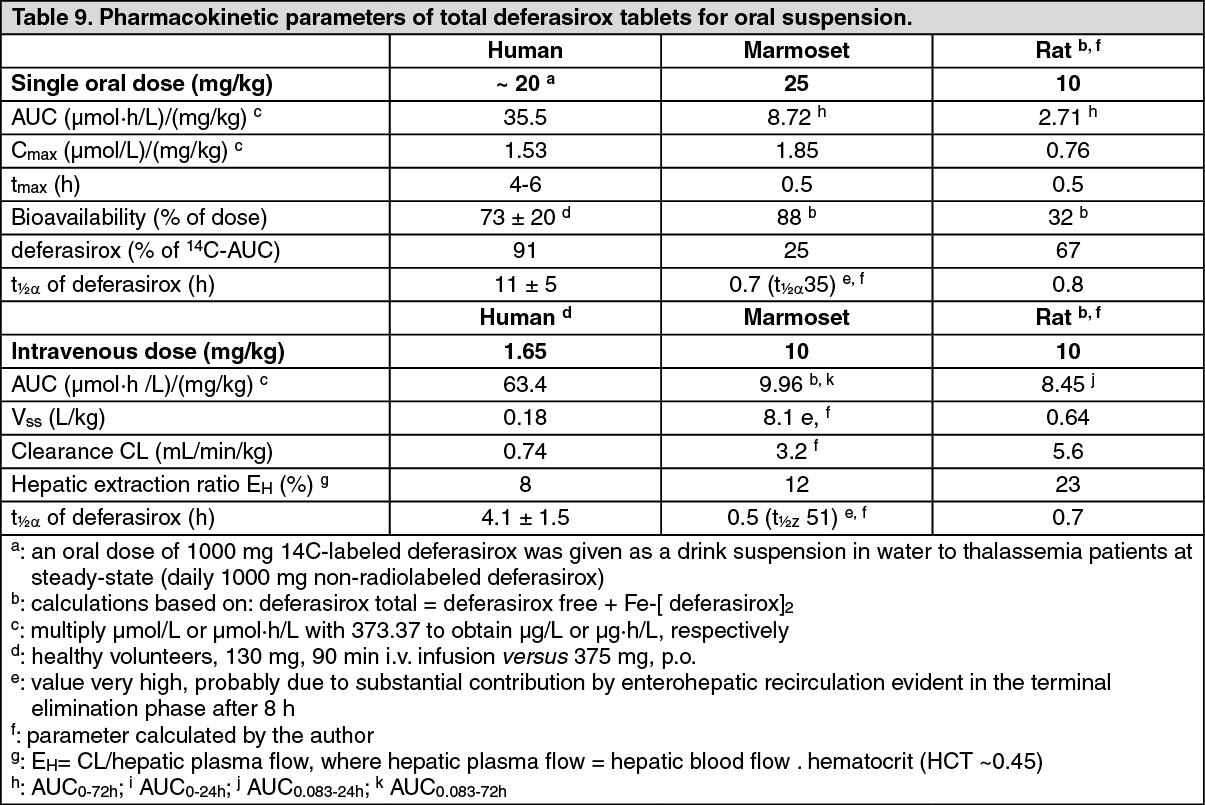 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image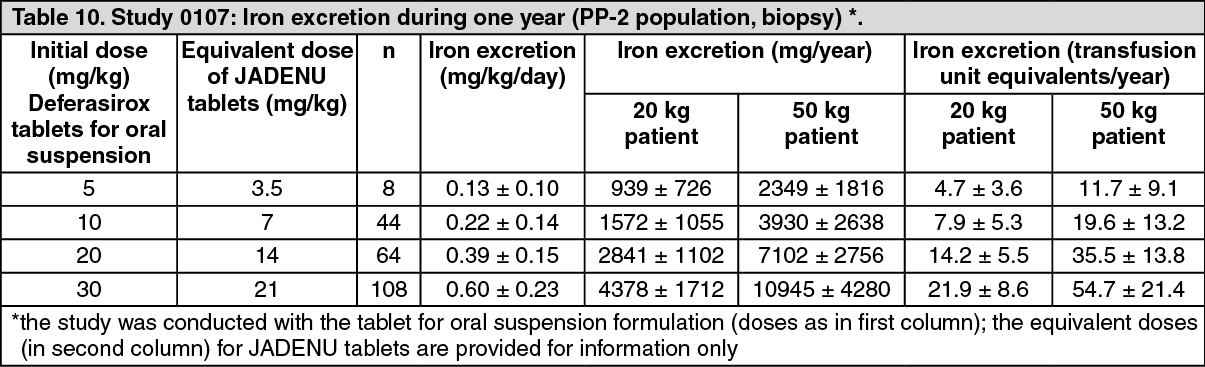 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image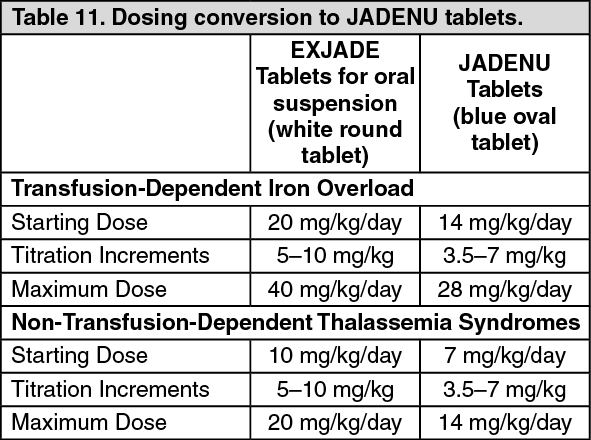 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image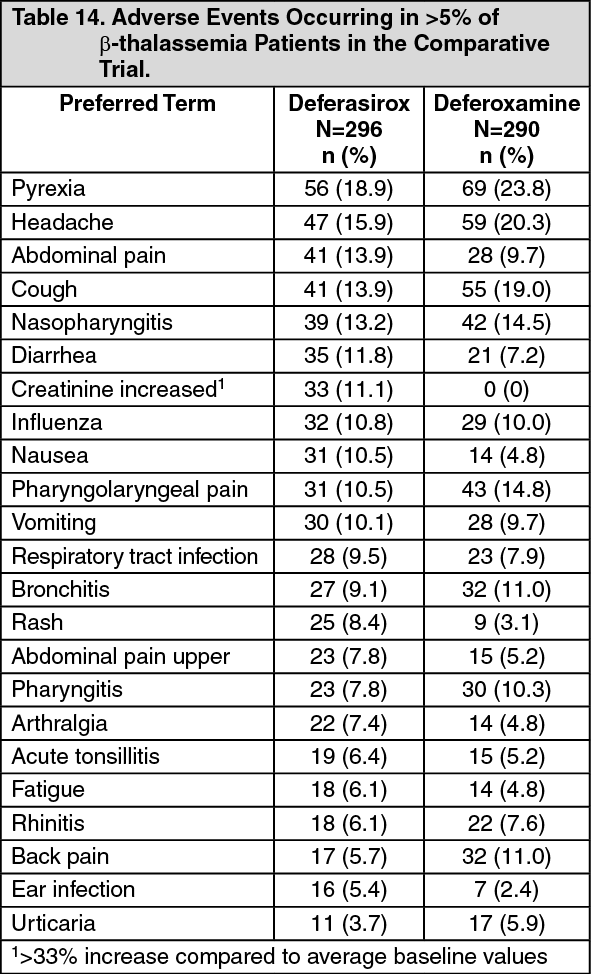 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image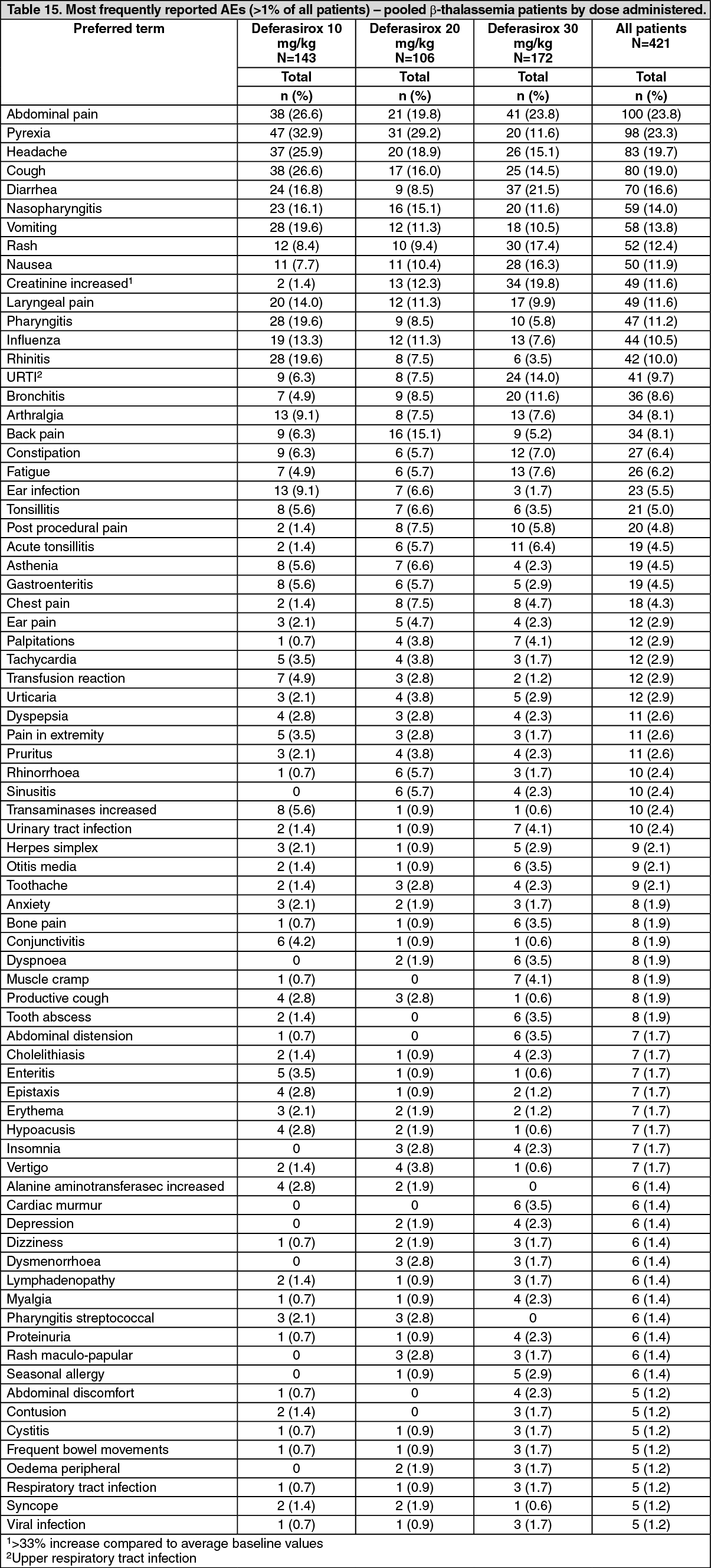 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
