ULTIVA should be administered only in a setting fully equipped for the monitoring and support of respiratory and cardiovascular function and by persons specifically trained in the use of anaesthetic drugs and the recognition and management of the expected adverse effects of potent opioids, including respiratory and cardiac resuscitation. Such training must include the establishment and maintenance of a patent airway and assisted ventilation.
Continuous infusions of ULTIVA must be administered by a calibrated infusion device into a fast-flowing i.v. line or via a dedicated i.v. line. This infusion line should be connected at, or close to, the venous cannula and primed, to minimise the potential dead space (see Instructions for Use/Handling under Cautions for Usage for additional information, including tables with examples of infusion rates by body weight to help titrate ULTIVA to the patient's anaesthetic needs).
Care should be taken to avoid obstruction or disconnection of infusion lines and to adequately clear the lines to remove residual ULTIVA after use (see Precautions).
ULTIVA is for i.v. use only and must not be administered by epidural or intrathecal injection (see Contraindications).
ULTIVA for injection is stable for 24 hours at room temperature (25°C) after reconstitution and further dilution to 20 to 250 micrograms/ml (50 micrograms/ml is the recommended dilution for adults and 20 to 25 micrograms/ml for paediatric patients aged 1 year and over) with one of the following i.v. fluids listed as follows: sterilised water for injections; 5% dextrose injection; 5% dextrose and 0.9% sodium chloride injection; 0.9% sodium chloride injection; 0.45% sodium chloride injection.
(see
Instructions for Use/Handling under Cautions for Usage for additional information, including tables to help titrate ULTIVA to the patient's anaesthetic needs).
The administration of ULTIVA during general anaesthesia must be individualised based on the patient's response. It is not recommended for use as the sole agent in general anaesthesia.
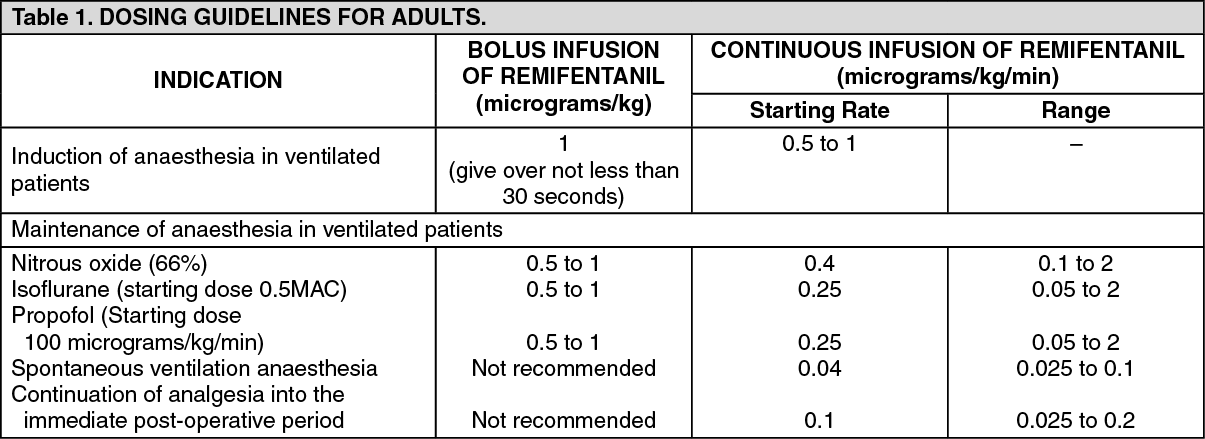
GENERAL ANAESTHESIA: Adults: The following table summarises the starting infusion rates and dose range. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
When given by bolus infusion at induction, ULTIVA should be administered over not less than 30 seconds.
At the doses recommended previously, ULTIVA significantly reduces the amount of hypnotic agent required to maintain anaesthesia. Therefore, isoflurane and propofol should be administered as recommended previously to avoid excessive depth of anaesthesia (see Concomitant medication as follows). No data are available for dosage recommendations for simultaneous use of other hypnotics with ULTIVA.
Induction of anaesthesia: ULTIVA should be administered with a hypnotic agent, such as propofol, thiopentone, or isoflurane, for the induction of anaesthesia. ULTIVA can be administered at an infusion rate of 0.5 to 1 micrograms/kg/min with or without an initial bolus infusion of 1 microgram/kg over not less than 30 seconds. If endotracheal intubation is to occur more than 8 to 10 minutes after the start of the infusion of ULTIVA, then a bolus infusion is not necessary.
Maintenance of anaesthesia: After endotracheal intubation, the infusion rate of ULTIVA should be decreased, according to anaesthetic technique, as indicated in the previous table. Due to the fast onset and short duration of action of remifentanil, the rate of administration during anaesthesia can be titrated upward in 25% to 100% increments or downward in 25% to 50% decrements, every 2 to 5 minutes to attain the desired level of mu-opioid response. In response to light anaesthesia, supplemental bolus infusions may be administered every 2 to 5 minutes.
Anaesthesia in spontaneously breathing anaesthetised patients with a secured airway (e.g. laryngeal mask anaesthesia): In spontaneously breathing anaesthetised patients with a secured airway, respiratory depression is likely to occur. Special care is needed to adjust the dose to the patient requirements and ventilatory support may be required. The recommended starting infusion rate for supplemental analgesia in spontaneously breathing anaesthetised patients is 0.04 micrograms/kg/min with titration to effect.
A range of infusion rates from 0.025 to 0.1 micrograms/kg/min has been studied. Bolus injections are not recommended in spontaneously breathing anaesthetised patients.
Continuation into the immediate post-operative period: In the event that longer acting analgesia has not been established prior to the end of surgery, ULTIVA may need to be continued to maintain analgesia during the immediate post-operative period until longer acting analgesia has reached its maximum effect.
In ventilated patients, the infusion rate should continue to be titrated to effect.
In patients who are breathing spontaneously, the infusion rate of ULTIVA should initially be decreased to a rate of 0.1 micrograms/kg/min. The infusion rate may then be increased or decreased by not greater than 0.025 micrograms/kg/min every 5 minutes, to balance the patient's level of analgesia and respiratory rate. ULTIVA should only be used in a setting fully equipped for the monitoring and support of respiratory and cardiovascular function, under the close supervision of persons specifically trained in the recognition and management of the respiratory effects of potent opioids.
The use of bolus injections of ULTIVA to treat pain during the post-operative period is not recommended in patients who are breathing spontaneously.
Concomitant medication: ULTIVA decreases the amounts or doses of inhaled anaesthetics, hypnotics and benzodiazepines required for anaesthesia (see Interactions).
Doses of the following agents used in anaesthesia, isoflurane, thiopentone, propofol and temazepam have been reduced by up to 75% when used concurrently with ULTIVA.
Guidelines for discontinuation: Due to the very rapid offset of action of ULTIVA, no residual opioid activity will be present within 5 to 10 minutes after discontinuation. For those patients undergoing surgical procedures where post-operative pain is anticipated, analgesics should be administered prior to or immediately following discontinuation of ULTIVA. Sufficient time must be allowed to reach the maximum effect of the longer acting analgesic. The choice of analgesic should be appropriate for the patient's surgical procedure and the level of post-operative care.
Paediatric Patients (1 to 12 years of age): Induction of anaesthesia: There are insufficient data to make a dosage recommendation.
Maintenance of anaesthesia: (See Table 2.)
Click on icon to see table/diagram/image
When given by bolus infusion, ULTIVA should be administered over not less than 30 seconds. Surgery should not commence until at least 5 minutes after the start of the ULTIVA infusion, if a simultaneous bolus dose has not been given. Paediatric patients should be monitored and the dose titrated to the depth of analgesia appropriate for the surgical procedure.
Concomitant medication: At the doses recommended previously, ULTIVA significantly reduces the amount of hypnotic agent required to maintain anaesthesia. Therefore, isoflurane, halothane and sevoflurane should be administered as recommended previously to avoid excessive depth of anaesthesia. No data are available for dosage recommendations for simultaneous use of other hypnotics with ULTIVA (see previously mentioned in General Anaesthesia: Adults: Concomitant medication).
Guidelines for discontinuation: Following discontinuation of the infusion, the offset of analgesic effect of ULTIVA is rapid and similar to that seen in adult patients. Appropriate post-operative analgesic requirements should be anticipated and implemented (see previously mentioned in General Anaesthesia: Adults: Guidelines for discontinuation).
Neonates/Infants (Aged less than 1 year): The pharmacokinetic profile of ULTIVA in neonates/infants (aged less than 1 year) is comparable to that seen in adults after correction for body weight differences. However, there are insufficient clinical data to make dosage recommendations for this age group.
CARDIAC ANAESTHESIA: Adults: (See Table 3.)
Click on icon to see table/diagram/image
Induction period of anaesthesia: After administration of hypnotic to achieve loss of consciousness, ULTIVA should be administered at an initial infusion rate of 1 microgram/kg/min. The use of bolus infusions of ULTIVA during induction in cardiac surgical patients is not recommended. Endotracheal intubation should not occur until at least 5 minutes after the start of the infusion.
Maintenance period of anaesthesia: After endotracheal intubation the infusion rate of ULTIVA should be titrated according to patient need. Supplemental bolus doses may also be given as required. High risk cardiac patients, such as those with poor ventricular function, should be administered a maximum bolus dose of 0.5 micrograms/kg. These dosing recommendations also apply during hypothermic cardiopulmonary bypass (see Pharmacology: Pharmacokinetics under Actions).
Concomitant medication: At the doses recommended previously, ULTIVA significantly reduces the amount of hypnotic agent required to maintain anaesthesia. Therefore, isoflurane and propofol should be administered as recommended previously to avoid excessive depth of anaesthesia. No data are available for dosage recommendations for simultaneous use of other hypnotics with ULTIVA (see previously mentioned in General Anaesthesia: Adults: Concomitant medication).
Continuation of post-operative analgesia prior to extubation: It is recommended that the infusion of ULTIVA should be maintained at the final intra-operative rate during transfer of patients to the post-operative care area. Upon arrival into this area the patient's level of analgesia and sedation should be closely monitored and the ULTIVA infusion rate adjusted to meet the individual patient's requirements.
Guidelines for discontinuation: Prior to discontinuation of ULTIVA, patients must be given alternative analgesic and sedative agents at a sufficient time in advance. The choice and dose of agent(s) should be appropriate for the patient's level of post-operative care (see previously mentioned in General Anaesthesia: Adults: Guidelines for discontinuation).
It is recommended that the ULTIVA infusion is discontinued by reducing the infusion rate by 25% decrements in at least 10 minutes intervals until the infusion is discontinued.
During weaning from the ventilator the ULTIVA infusion should not be increased and only down titration should occur, supplemented as required with alternative analgesics.
It is recommended that haemodynamic changes such as hypertension and tachycardia should be treated with alternative agents as appropriate.
Paediatric Patients: There are insufficient data to make a dosage recommendation for use during cardiac surgery.
USE IN INTENSIVE CARE: Adults: ULTIVA can be initially used alone for the provision of analgesia and sedation in mechanically ventilated intensive care patients.
It is recommended that ULTIVA is initiated at an infusion rate of 0.1 micrograms/kg/min to 0.15 micrograms/kg/min. The infusion rate should be titrated in increments of 0.025 micrograms/kg/min to achieve the desired level of analgesia and sedation. A period of at least 5 minutes should be allowed between dose adjustments. The level of analgesia and sedation should be carefully monitored, regularly reassessed and the ULTIVA infusion rate adjusted accordingly. If an infusion rate of 0.2 micrograms/kg/min is reached and the desired level of sedation is not achieved, it is recommended that dosing with an appropriate sedative agent is initiated. The dose of sedative agent should be titrated to obtain the desired level of sedation. Further increases to the ULTIVA infusion rate in increments of 0.025 micrograms/kg/min may be made if additional analgesia is required.
ULTIVA has been studied in intensive care patients in well controlled clinical trials for up to 3 days. There are limited additional clinical trial data for longer durations.
The following table summarises the starting infusion rates and typical dose range for provision of analgesia and sedation in individual patient: (See Table 4.)
Click on icon to see table/diagram/image
Bolus doses of ULTIVA are not recommended in the intensive care setting.
The use of ULTIVA will reduce the dosage requirement of any concomitant sedative agents.
Typical starting doses for sedative agents, if required, are given as follows. (See Table 5.)
Click on icon to see table/diagram/image
To allow separate titration of the respective agents sedative agents should not be prepared as one mixture in the same infusion bag.
Additional analgesia for ventilated patients undergoing stimulating procedures: An increase in the existing ULTIVA infusion rate may be required to provide additional analgesic cover for ventilated patients undergoing stimulating and/or painful procedures such as endotracheal suctioning, wound dressing and physiotherapy. It is recommended that an ULTIVA infusion rate of at least 0.1 micrograms/kg/min should be maintained for at least 5 minutes prior to the start of the stimulating procedure. Further dose adjustments may be made every 2 to 5 minutes in increments of 25% to 50% in anticipation of, or in response to, additional requirement for analgesia. A mean infusion rate of 0.25 micrograms/kg/min, maximum 0.75 micrograms/kg/min, has been administered for provision of additional anaesthesia during stimulating procedures.
Guidelines for discontinuation: Prior to discontinuation of ULTIVA, patients must be given alternative analgesic and sedative agents at a sufficient time in advance. The appropriate choice and dose of agent(s) should be anticipated and implemented.
In order to ensure a smooth emergence from a remifentanil-based regimen it is recommended that the infusion rate of ULTIVA is titrated in stages to 0.1 micrograms/kg/min over a period up to 1 hour prior to extubation.
Following extubation, the infusion rate should be reduced by 25% decrements in at least 10 minutes intervals until the infusion is discontinued. During weaning from the ventilator the ULTIVA infusion should not be increased and only down titration should occur, supplemented as required with alternative analgesics.
Paediatric Intensive Care Patients: There are no data available on use in paediatric patients.
OTHER POPULATIONS: Elderly (over 65 years of age): GENERAL ANAESTHESIA: The initial starting dose of ULTIVA administered to patients over 65 should be half the recommended adult dose and then titrated to individual patient need as an increased sensitivity to the pharmacological effects of ULTIVA has been seen in this patient population.
This dose adjustment applies to use in all phases of anaesthesia including induction, maintenance and immediate post-operative analgesia.
CARDIAC ANAESTHESIA: No initial dose reduction is required (see Table 3: Cardiac Anaesthesia: Dosing guidelines as previously mentioned).
INTENSIVE CARE: No initial dose reduction is required (see Use in Intensive Care as previously mentioned).
Obese patients: It is recommended that for obese patients the dosage of ULTIVA should be reduced and based upon ideal body weight as the clearance and volume of distribution of remifentanil are better correlated with ideal body weight than actual body weight in this population.
Renal impairment: No dosage adjustment relative to that used in healthy adults is necessary in renally impaired patients, including those undergoing renal replacement therapy, as the pharmacokinetic profile of ULTIVA is unchanged in this patient population.
Hepatic impairment: No adjustment of the initial dose, relative to that used in healthy adults, is necessary as the pharmacokinetic profile of ULTIVA is unchanged in this patient population. However, patients with severe hepatic impairment may be slightly more sensitive to the respiratory depressant effects of remifentanil. These patients should be closely monitored and the dose of ULTIVA titrated to individual patient need.
Neurosurgery: Limited clinical experience in patients undergoing neurosurgery has shown that no special dosage recommendations are required.
ASA III/IV patients: GENERAL ANAESTHESIA: As the haemodynamic effects of potent opioids can be expected to be more pronounced in ASA III/IV patients, caution should be exercised in the administration of ULTIVA in this population. Initial dosage reduction and subsequent titration to effect is therefore recommended.
CARDIAC ANAESTHESIA: No initial dose reduction is required (see Table 3: Cardiac Anaesthesia: Dosing guidelines as previously mentioned).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image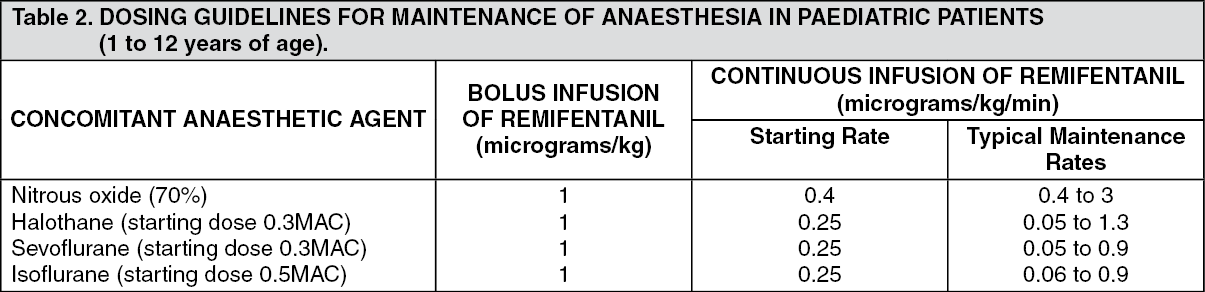 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image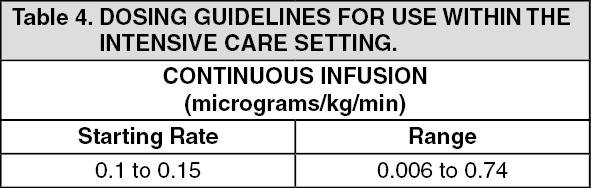 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image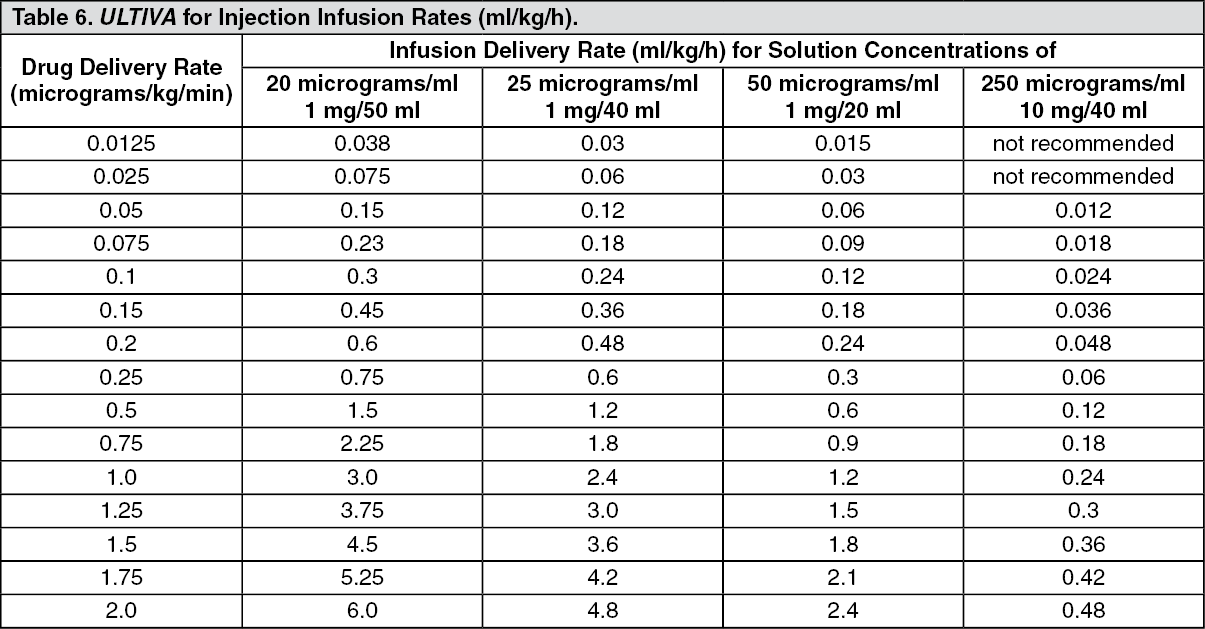 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image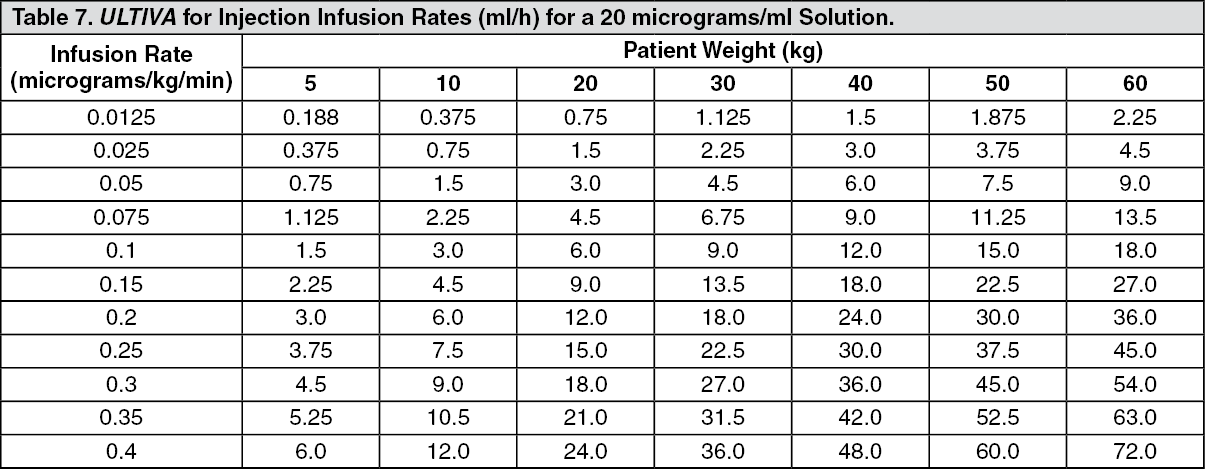 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image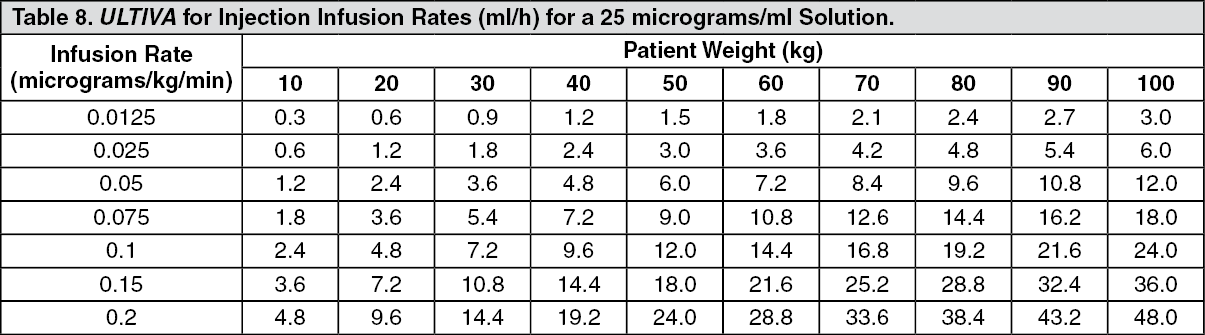 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image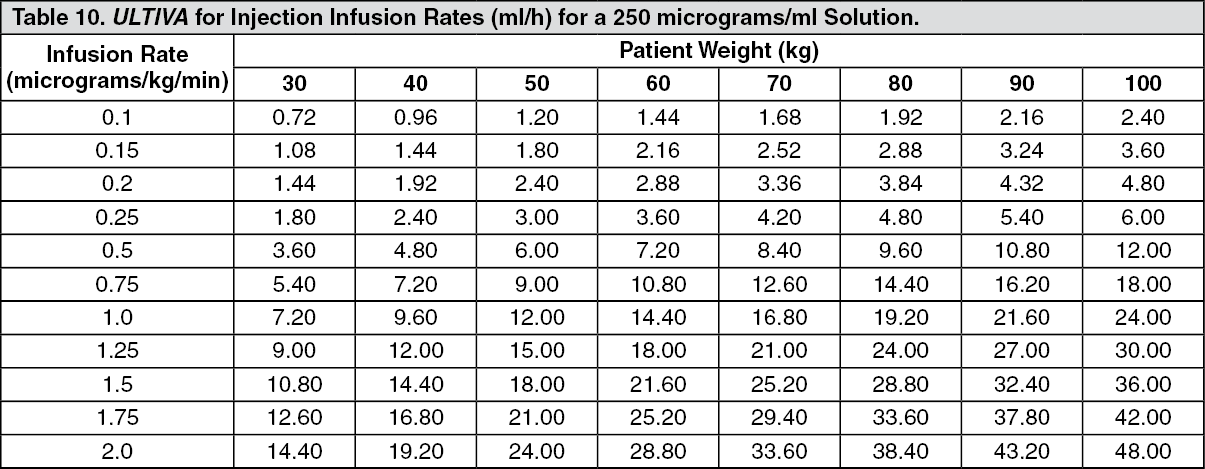 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
