Syrup: RETROVIR therapy should be initiated by a physician experienced in the management of HIV infection.
Adults and adolescents weighing at least 30kg: The recommended dose of RETROVIR in combination with other anti-retroviral agents is 250 or 300 mg twice daily.
Children: Oral solution/syrup: Children weighing at least 9 kg and less than 30 kg: The recommended dose of RETROVIR is 0.9 mL/kg (9 mg/kg) twice daily in combination with other anti-retroviral agents (e.g. a 15 kg child would require a 13.5 mL dose or oral solution twice daily). The maximum dosage should not exceed 300 mg (30 mL) twice daily.
Children weighing at least 4 kg and less than 9 kg: The recommended dose of RETROVIR is 1.2 mL/kg (12 mg/kg) twice daily in combination with other antiretroviral agents (e.g. a 5 kg neonate would require a 6 mL dose of oral solution twice daily).
Available data are insufficient to propose specific dosage recommendations for children weighing less than 4kg (see as follows - maternal foetal transmission and Pharmacology: Pharmacokinetics under Actions).
Elderly: Zidovudine pharmacokinetics have not been studied in patients over 65 years of age and no specific data are available. However, since special care is advised in this age group due to age-associated changes such as the decrease in renal function and alterations in haematological parameters, appropriate monitoring of patients before and during use of RETROVIR is advised.
Renal impairment: In patients with severe renal impairment daily dosages of 300 to 400 mg should be appropriate. Haematological parameters and clinical response may influence the need for subsequent dosage adjustment. Haemodialysis and peritoneal dialysis have no significant effect on RETROVIR elimination whereas elimination of the glucuronide metabolite is increased. For patients with end-stage renal disease maintained on haemodialysis or peritoneal dialysis, the recommended dose is 100 mg every 6 to 8 hours (see Pharmacology: Pharmacokinetics under Actions).
Hepatic impairment: Data in patients with cirrhosis suggest that accumulation of zidovudine may occur in patients with hepatic impairment because of decreased glucuronidation. Dosage adjustments may be necessary, but as there is only limited data available precise recommendations cannot be made. If monitoring of plasma zidovudine levels is not feasible, physicians will need to monitor for signs of intolerance and adjust the dose and/or increase the interval between doses as appropriate.
Dosage adjustments in patients with haematological adverse reactions: Dosage reduction or interruption of RETROVIR therapy may be necessary in patients whose haemoglobin level falls to between 7.5 g/dl (4.65 mmol/l) and 9 g/dl (5.59 mmol/l) or whose neutrophil count falls to between 0.75 x 10
9/l and 1.0 x 10
9/l (see Contraindications and Precautions).
Dosage in the prevention of maternal-foetal transmission: The following RETROVIR dosage regimens have been shown to be effective (see Use in Pregnancy & Lactation).
ACTG 076 study: The recommended dose of RETROVIR for pregnant women (over 14 weeks of gestation) is 500 mg/day orally (100 mg five times daily) until the beginning of labour. During labour and delivery RETROVIR should be administered intravenously at 2 mg/kg bodyweight given over 1 hour, followed by a continuous intravenous infusion at 1 mg/kg/h until the umbilical cord is clamped.
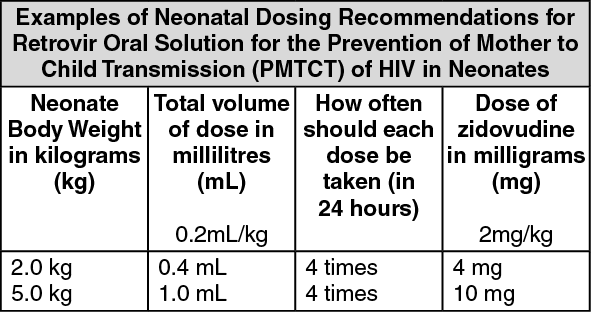
Neonates should be given RETROVIR 0.2 mL/kg (2 mg/kg) bodyweight of oral solution every 6 hours starting within 12 hours after birth, and continuing until 6 weeks old.
An appropriate sized syringe with 0.1 mL graduation should be used to ensure accurate dosing of neonates.
(See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Infants unable to receive oral dosing should be given RETROVIR infusion intravenously at 1.5 mg/kg bodyweight infused over 30 minutes every 6 hours.
IV infusion: RETROVIR therapy should be initiated by a physician experienced in the management of HIV infection.
The required dose of RETROVIR I.V. for Infusion must be administered by slow intravenous infusion of the diluted product over a one-hour period.
RETROVIR I.V. for Infusion must NOT be given intramuscularly.
Patients should receive RETROVIR I.V. for Infusion only until oral therapy can be administered.
Dilution: RETROVIR I.V. for Infusion must be diluted prior to administration (see Instructions for Use/Handling under Cautions for Usage).
Adults and adolescents over 12 years of age: A dosage of RETROVIR I.V. for Infusion of 1 or 2 mg zidovudine/kg every four hours provides similar exposure (AUC) to an oral dosage of 1.5 or 3 mg zidovudine/kg every four hours (600 or 1200 mg/day for a 70 kg patient).
Children (3 months to 12 years): Limited data are available on the use of RETROVIR I.V. for Infusion in children. A range of dosages between 80 and 160 mg/m
2 body surface area every 6 hours (320 to 640 mg/m
2/day) have been used. However, estimated exposure following doses of between 240 to 320 mg/m
2 per day in 3 or 4 divided doses would approximately correspond to the currently recommended oral dose of 360 to 480 mg/m
2 per day although there is no efficacy data currently available on these lower intravenous doses.
Children (less than 3 months): Available data are insufficient to propose specific dosage recommendations (see as follows - maternal foetal transmission and Pharmacology: Pharmacokinetics under Actions).
Elderly: Zidovudine pharmacokinetics have not been studied in patients over 65 years of age and no specific data are available. However, since special care is advised in this age group due to age-associated changes such as the decrease in renal function and alterations in haematological parameters, appropriate monitoring of patients before and during use of RETROVIR is advised.
Renal impairment: In patients with severe renal impairment, the recommended intravenous dosage is 1 mg/kg 3-4 times daily. This is equivalent to the current recommended oral daily dosage for this patient group of 300-400 mg allowing for oral bioavailability of 60-70%. Haematological parameters and clinical response may influence the need for subsequent dosage adjustment. Haemodialysis and peritoneal dialysis have no significant effect on zidovudine elimination whereas elimination of the glucuronide metabolite is increased. For patients with end-stage renal disease maintained on haemodialysis or peritoneal dialysis, the recommended dose is 100 mg every 6 to 8 hours (see Pharmacology: Pharmacokinetics under Actions).
Hepatic impairment: Data in patients with cirrhosis suggest that accumulation of zidovudine may occur in patients with hepatic impairment because of decreased glucuronidation. Dosage adjustments may be necessary, but as there is only limited data available precise recommendations cannot be made. If monitoring of plasma zidovudine levels is not feasible, physicians will need to monitor for signs of intolerance and adjust the dose and/or increase the interval between doses as appropriate.
Dosage adjustments in patients with haematological adverse reactions: Dosage reduction or interruption of RETROVIR therapy may be necessary in patients whose haemoglobin level falls to between 7.5 g/dl (4.65 mmol/l) and 9 g/dl (5.59 mmol/l) or whose neutrophil count falls to between 0.75 x 10
9/l and 1.0 x 10
9/l (see Contraindications and Precautions).
Dosage in the prevention of maternal-foetal transmission: The following dosage regimen has been shown to be effective. Pregnant women (over 14 weeks of gestation) should be given 500 mg/day orally (100 mg five times/daily) until the beginning of labour. During labour and delivery RETROVIR should be administered intravenously at 2 mg/kg bodyweight given over 1 hour, followed by a continuous intravenous infusion at 1 mg/kg/h until the umbilical cord is clamped.
The newborn infants should be given RETROVIR 2 mg/kg bodyweight of oral solution every 6 hours starting within 12 hours after birth and continuing until 6 weeks old. An appropriate sized syringe should be used to ensure accurate dosing of infants. Infants unable to receive oral dosing should be given RETROVIR infusion intravenously at 1.5 mg/kg bodyweight infused over 30 minutes every 6 hours.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
