Pharmacology: Mechanism of Action: Evolocumab is a human monoclonal IgG2 directed against human proprotein convertase subtilisin kexin type 9 (PCSK9). PCSK9 binds to the low-density lipoprotein (LDL) receptor (LDLR) on the surface of hepatocytes to promote LDLR degradation within the liver. By inhibiting the binding of PCSK9 to LDLR, evolocumab increases the number of LDLRs available to clear LDL from the blood, thereby lowering LDL-C levels.
Pharmacodynamics: Following single subcutaneous administration of 140 mg or 420 mg of evolocumab, maximum suppression of circulating unbound PCSK9 occurred by 4 hours. Unbound PCSK9 concentrations returned toward baseline when evolocumab concentrations decreased below the limit of quantitation.
Clinical Studies: Adult Patients with Established Cardiovascular Disease: FOURIER (NCT01764633) was a double-blind, randomized, placebo-controlled, event-driven trial in 27,564 (13,784 REPATHA, 13,780 placebo) adult patients with established cardiovascular disease and with LDL-C ≥70 mg/dL and/or non-HDL-C ≥100 mg/dL despite high- or moderate-intensity statin therapy. Patients were randomly assigned 1:1 to receive either subcutaneous injections of REPATHA (140 mg every 2 weeks or 420 mg once monthly) or placebo; 86% used the every-2-week regimen throughout the trial. The median follow-up duration was 26 months. Overall, 99.2% of patients were followed until the end of the trial or death.
The mean (SD) age at baseline was 63 (9) years, with 45% being at least 65 years old; 25% were women. The trial population was 85% White, 2% Black, and 10% Asian; 8% identified as Hispanic ethnicity. Regarding prior diagnoses of cardiovascular disease, 81% had prior myocardial infarction, 19% prior non-hemorrhagic stroke, and 13% had symptomatic peripheral arterial disease. Selected additional baseline risk factors included hypertension (80%), diabetes mellitus (1% type 1; 36% type 2), current daily cigarette smoking (28%), New York Heart Association class I or II congestive heart failure (23%), and eGFR <60 mL/min per 1.73 m
2 (6%). Most patients were on a high- (69%) or moderate-intensity (30%) statin therapy at baseline, and 5% were also taking ezetimibe. Most patients were taking at least one other cardiovascular medication including anti-platelet agents (93%), beta blockers (76%), angiotensin converting enzyme (ACE) inhibitors (56%), or angiotensin receptor blockers (23%). On stable background lipid-lowering therapy, the median [Q1, Q3] LDL-C at baseline was 92 [80, 109] mg/dL; the mean (SD) was 98 (28) mg/dL.
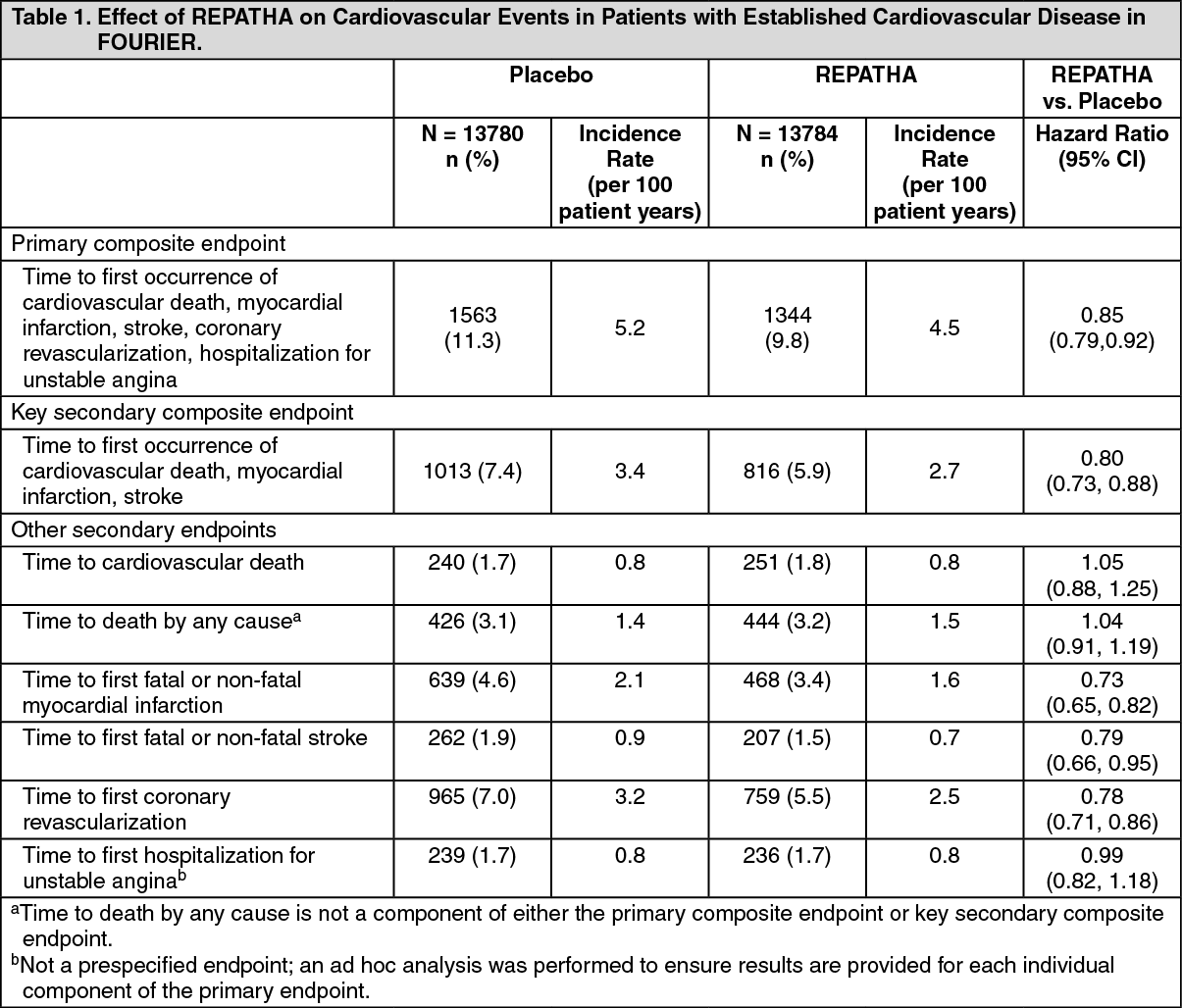
REPATHA significantly reduced the risk for the primary composite endpoint (time to first occurrence of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization; p <0.0001) and the key secondary composite endpoint (time to first occurrence of cardiovascular death, myocardial infarction, or stroke; p <0.0001). The Kaplan-Meier estimates of the cumulative incidence of the primary and key secondary composite endpoints over time are shown in Figure 1 and Figure 2 as follows.
The results of primary and secondary efficacy endpoints are shown in Table 1 as follows. (See Table 1, Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was -63% (95% CI: -63%, -62%) and from baseline to Week 72 was -57% (95% CI: -58%, -56%). At Week 48, the median [Q1, Q3] LDL-C was 26 [15, 46] mg/dL in the REPATHA group, with 47% of patients having LDL-C <25 mg/dL.
In EBBINGHAUS (NCT02207634), a substudy of 1974 patients enrolled in the FOURIER trial, REPATHA was non-inferior to placebo on selected cognitive function domains as assessed with the use of neuropsychological function tests over a median follow-up of 19 months.
FOURIER-OLE (study 1 and study 2) consisted of two open-label, single-arm, multicenter, extension studies to evaluate the long-term safety, tolerability, and efficacy of REPATHA in patients with established cardiovascular disease who completed the FOURIER study. Enrolled patients received REPATHA 140 mg every 2 weeks or 420 mg once monthly for approximately 5 years and continued moderate- (22.2%) or high-intensity (74.8%) background statin therapy. Of the 5031 patients who received at least one dose of REPATHA in study 1, 2499 patients received REPATHA and 2532 patients received placebo in the FOURIER study. Of the 1599 patients who received at least one dose of REPATHA in study 2, 854 patients received REPATHA and 745 patients received placebo in the FOURIER study. Upon completion of study 1 and study 2, patients randomized to REPATHA in the FOURIER study had up to 8.4 years (median 85.4 months) and 8.0 years of total REPATHA exposure (median 80.2 months) and patients randomized to placebo had up to 5.25 years (median 60.0 months) and 4.9 years of total REPATHA exposure (median 55.1 months), respectively.
In study 1 and 2 combined, 72.4% (n=4802) of patients achieved a lowest post-baseline LDL-C <25 mg/dL (0.65 mmol/L), 87.0% (n=5765) of patients achieved an LDL-C <40 mg/dL (1.03 mmol/L), and 11.9% (n=792) of patients had an all post-baseline LDL-C ≥40 mg/dL (1.03 mmol/L). Of the patients who achieved post-baseline low LDL-C (<25 mg/dL or <40 mg/dL), the overall subject incidences of treatment emergent adverse events were 80.0% patients who achieved LDL-C <25 mg/dL and 82.7% in patients who achieved LDL-C <40 mg/dL compared to 85.0% in patients with LDL-C ≥40 mg/dL. The overall subject incidences of serious treatment emergent adverse events were 37.7% in patients who achieved LDL-C <25 mg/dL and 40.0% in patients who achieved LDL-C <40 mg/dL compared to 41.5% in patients with LDL-C ≥40 mg/dL.
The mean percent reduction from baseline in LDL-C was stable during the OLE study period and ranged from 53.4% to 59.1% for study 1 and 62.5% to 67.2% for study 2, regardless of the patient's original randomised treatment group in the FOURIER study. This appears to translate into a numerically lower subject incidence rate of adjudicated exploratory CV endpoints of the composite of CV death, MI and stroke for patients who had received REPATHA in both the FOURIER and FOURIER-OLE studies compared with patients who had received placebo in the FOURIER study and REPATHA in the FOURIER-OLE studies.
Overall, no new safety findings were identified in these studies.
Primary Hyperlipidemia (Including Heterozygous Familial Hypercholesterolemia): LAPLACE-2 (NCT01763866) was a multicenter, double-blind, randomized controlled 12-week trial in which patients were initially randomized to an open-label specific statin regimen for a 4-week lipid stabilization period followed by random assignment to subcutaneous injections of REPATHA 140 mg every 2 weeks, REPATHA 420 mg once monthly, or placebo for 12 weeks. The trial included 1896 patients with hyperlipidemia who received REPATHA, placebo, or ezetimibe as add-on therapy to daily doses of statins (atorvastatin, rosuvastatin, or simvastatin). Ezetimibe was also included as an active control only among those assigned to background atorvastatin. Overall, the mean age at baseline was 60 years (range: 20 to 80 years), 35% were ≥65 years old, 46% women, 94% White, 4% were Black, and 1% Asian; 5% identified as Hispanic or Latino ethnicity. After 4 weeks of background statin therapy, the mean baseline LDL-C ranged between 77 and 127 mg/dL across the five background therapy arms.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was -71% (95% CI: -74%, -67%; p <0.0001) and -63% (95% CI: -68%, -57%; p ˂0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. The difference between REPATHA and ezetimibe in mean percent change in LDL-C from baseline to Week 12 was -45% (95% CI: -52%, -39%; p <0.0001) and -41% (95% CI: -47%, -35%; p ˂0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results (see Table 2 and Figure 3).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
DESCARTES (NCT01516879) was a multicenter, double-blind, randomized, placebo-controlled, 52-week trial that included 901 patients with hyperlipidemia who received protocol-determined background lipid-lowering therapy of a cholesterol-lowering diet either alone or in addition to atorvastatin (10 mg or 80 mg daily) or the combination of atorvastatin 80 mg daily with ezetimibe. After stabilization on background therapy, patients were randomly assigned to the addition of placebo or REPATHA 420 mg administered subcutaneously once monthly. Overall, the mean age at baseline was 56 years (range: 25 to 75 years), 23% were ≥65 years, 52% women, 80% White, 8% Black, and 6% Asian; 6% identified as Hispanic or Latino ethnicity. After stabilization on the assigned background therapy, the mean baseline LDL-C ranged between 90 and 117 mg/dL across the four background therapy groups.
In these patients with hyperlipidemia on a protocol-determined background therapy, the difference between REPATHA 420 mg once monthly and placebo in mean percent change in LDL-C from baseline to Week 52 was -55% (95% CI: -60%, -50%; p ˂0.0001) (Table 3 and Figure 4). For additional results see Table 3. (See Table 3 and Figure 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
MENDEL-2 (NCT01763827) was a multicenter, double-blind, randomized, placebo- and active-controlled, 12-week trial that included 614 patients with hyperlipidemia who were not taking lipid-lowering therapy at baseline. Patients were randomly assigned to receive subcutaneous injections of REPATHA 140 mg every 2 weeks, REPATHA 420 mg once monthly, or placebo for 12 weeks. Blinded administration of ezetimibe was also included as an active control. Overall, the mean age at baseline was 53 years (range: 20 to 80 years), 18% were ≥65 years old, 66% were women, 83% White, 7% Black, and 9% Asian; 11% identified as Hispanic or Latino ethnicity. The mean baseline LDL-C was 143 mg/dL.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was -55% (95% CI: -60%, -50%; p <0.0001) and -57% (95% CI: -61%, -52%; p ˂0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. The difference between REPATHA and ezetimibe in mean percent change in LDL-C from baseline to Week 12 was -37% (95% CI: -42%, -32%; p <0.0001) and -38% (95% CI: -42%, -34%; p ˂0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results (see Table 4).
Click on icon to see table/diagram/image
RUTHERFORD-2 (NCT01763918) was a multicenter, double-blind, randomized, placebo-controlled, 12-week trial in 329 patients with heterozygous familial hypercholesterolemia (HeFH) on statins with or without other lipid-lowering therapies. Patients were randomized to receive subcutaneous injections of REPATHA 140 mg every two weeks, 420 mg once monthly, or placebo. HeFH was diagnosed by the Simon Broome criteria (1991). 38% of patients had clinical atherosclerotic cardiovascular disease. The mean age at baseline was 51 years (range: 19 to 79 years), 15% of the patients were ≥65 years old, 42% were women, 90% were White, 5% were Asian, and 1% were Black. The average LDL-C at baseline was 156 mg/dL with 76% of the patients on high-intensity statin therapy.
The differences between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was -61% (95% CI: -67%, -55%; p <0.0001) and -60% (95% CI: -68%, -52%; p <0.0001) for the 140 mg every 2 weeks and 420 mg once monthly dosages, respectively. For additional results (see Table 5 and Figure 5).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
OSLER-1 was a multicenter, randomized, controlled, open-label, 5-year extension study to assess the long-term safety and efficacy of Repatha in patients with hyperlipidemia. A total of 1324 patients who completed treatment in 1 of 5 parent (Phase 2) studies enrolled in the study. Patients were randomized 2:1 to receive either Repatha 420 mg once monthly plus standard of care (evolocumab group) or standard of care alone (control group) for the first year of the study (year 1). Year 1 of the study was controlled. At the end of the first year, patients entered the all evolocumab period (year 2+) in which all patients received open-label Repatha for up to an approximately additional 4 years. Repatha 420 mg once monthly significantly reduced LDL-C from baseline at week 12 and week 52 compared with control (nominal p <0.001). Treatment effects were maintained over 272 weeks as demonstrated by reduction in LDL-C from week 12 in the parent study to week 260 in the open-label extension (Figure 5). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG, and Lp(a), and increased HDL-C and ApoA1 from baseline to week 52 compared with control (nominal p <0.001). LDL-C and other lipid parameters returned to baseline within 12 weeks after discontinuation of Repatha at beginning of OSLER-1 without evidence of rebound (see Figure 6).
Click on icon to see table/diagram/image
OSLER-2 was a multicenter, randomized, controlled, open-label, 3-year extension study designed to assess the long-term safety and efficacy of Repatha in patients with hypercholesterolemia. A total of 3681 patients who completed treatment in 1 of 9 parent (Phase 3) studies enrolled in the study. Patients were randomized 2:1 to receive either Repatha plus standard of care (evolocumab group) or standard of care alone (control group) for the first year of the study (year 1). Year 1 of the study was controlled. At the end of the first year, patients entered the all evolocumab period (year 2) in which all patients received open-label Repatha for up to two more years. Repatha significantly reduced LDL-C from baseline at week 12 and week 48 compared with control (nominal p <0.001). Treatment effects were maintained as demonstrated by reduction in LDL-C from week 12 to week 104 in the open-label extension (Figure 6). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG, and Lp(a), and increased HDL-C and ApoA1 from baseline to week 48 compared with control (nominal p <0.001). LDL-C and other lipid parameters returned to baseline within 12 weeks after discontinuation of Repatha without evidence of rebound (see Figure 7).
Click on icon to see table/diagram/image
TAUSSIG was a multicenter, open-label 5-year extension study to assess the long-term safety and efficacy of Repatha in patients with severe familial hypercholesterolemia (FH), including HoFH, who were treated with Repatha as an adjunct to other lipid-lowering therapies. A total of 194 severe FH (non-HoFH) patients and 106 HoFH patients enrolled in TAUSSIG. All patients in the study were initially treated with Repatha 420 mg once monthly except for those receiving lipid apheresis at enrollment, who began with Repatha 420 mg every 2 weeks. Dose frequency in non-apheresis patients could be titrated up to 420 mg once every 2 weeks based on LDL-C response and PCSK9 levels. Long-term use of Repatha demonstrated a sustained treatment effect as evidenced by reduction of LDL-C in patients with severe FH (non-HoFH) (Table 6). Changes in other lipid parameters (TC, ApoB, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a sustained effect of long-term Repatha administration in patients with severe FH (non-HoFH). (See Table 6.)
Click on icon to see table/diagram/image
Pediatric Patients with HeFH: HAUSER-RCT (NCT02392559) was a randomized, multicenter, placebo-controlled, double-blind, 24-week trial in 157 pediatric patients aged 10 to 17 years with HeFH [see Use in Children under Precautions]. HeFH was diagnosed by diagnostic criteria for HeFH [Simon Broome Register Group (1991), the Dutch Lipid Clinic Network (1999), MEDPED (1993)] or by genetic testing. Patients were required to be on a low-fat diet and optimized background lipid-lowering therapy. Patients were randomly assigned 2:1 to receive 24 weeks of subcutaneous once monthly 420 mg REPATHA or placebo; 104 patients received REPATHA and 53 patients received placebo. The mean age was 14 years (range: 10 to 17 years), 56% were female, 85% White, 1% Black, 1% Asian, 13% Other, and 8% Hispanic. The mean LDL-C at baseline was 184 mg/dL; 17% of patients were on high-intensity statin, 62% on moderate-intensity statin, and 13% on ezetimibe.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 24 was -38% (95% CI: -45%, -31%; p <0.0001). For additional results, see Table 7 and Figure 8. (See Figure 8 and Table 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Adult and Pediatric Patients with HoFH: TESLA (NCT01588496) was a multicenter, double-blind, randomized, placebo-controlled, 12-week trial in 49 patients (not on lipid-apheresis therapy) with homozygous familial hypercholesterolemia (HoFH). In this trial, 33 patients received subcutaneous injections of 420 mg of REPATHA once monthly and 16 patients received placebo as an adjunct to other lipid-lowering therapies (e.g., statins, ezetimibe). The mean age at baseline was 31 years, 49% were women, 90% White, 4% were Asian, and 6% other. The trial included 10 adolescents (ages 13 to 17 years), 7 of whom received REPATHA. The mean LDL-C at baseline was 349 mg/dL with all patients on statins (atorvastatin or rosuvastatin) and 92% on ezetimibe. The diagnosis of HoFH was made by genetic confirmation or a clinical diagnosis based on a history of an untreated LDL-C concentration >500 mg/dL together with either xanthoma before 10 years of age or evidence of HeFH in both parents.
The difference between REPATHA and placebo in mean percent change in LDL-C from baseline to Week 12 was -31% (95% CI: -44%, -18%; p <0.0001). For additional results see Table 8.
Patients known to have two LDL-receptor negative alleles (little to no residual function) did not respond to REPATHA. (See Table 8.)
Click on icon to see table/diagram/image
Long-term Efficacy in Homozygous Familial Hyperlipidemia: TAUSSIG (NCT01624142) was a multicenter, open-label 5-year extension study with REPATHA in 106 patients with HoFH, who were treated with REPATHA as an adjunct to other lipid-lowering therapies. The study included 14 pediatric patients (ages 13 to 17 years). All patients in the study were initially treated with REPATHA 420 mg once monthly except for those receiving lipid apheresis at enrollment, who began with REPATHA 420 mg every 2 weeks. Dose frequency in non-apheresis patients could be titrated up to 420 mg once every 2 weeks based on LDL-C response and PCSK9 levels.
A total of 48 patients with HoFH received REPATHA 420 mg once monthly for at least 12 weeks in followed by REPATHA 420 mg every 2 weeks for at least 12 weeks. Mean percent change from baseline in LDL-C were -20% at Week 12 of 420 mg once monthly treatment and -30% at Week 12 of 420 mg every 2 weeks treatment, based on available data.
HAUSER-OLE, NCT02624869 was an open-label, single-arm, multicenter, 80-week study to evaluate the safety, tolerability, and efficacy of REPATHA for LDL-C reduction in pediatric patients aged 10 to 17 years with HoFH [see Use in Children under Precautions]. Patients were on a low-fat diet and receiving background lipid-lowering therapy. Overall, 12 patients with HoFH received 420 mg REPATHA subcutaneously once monthly. The mean age was 12 years (range 11 to 17 years), 17% were female, 75% White, 17% Asian, and 8% Other. Median (Q1, Q3) LDL-C at baseline was 398 (343, 475) mg/dL, and all patients were on statins (atorvastatin or rosuvastatin) and ezetimibe. No patients were receiving lipid apheresis. The diagnosis of HoFH was made by genetic confirmation in all patients but enrollment by a clinical diagnosis was permitted. The median (Q1, Q3) percent change in LDL-C from baseline to Week 80 was -14% (-41, 4). Two of the 3 subjects with <5% LDLR activity responded to evolocumab treatment.
Regression of Atherosclerosis: GLAGOV was a phase 3, double-blind, randomized, placebo-controlled study to evaluate the effects of Repatha treatment on coronary atherosclerotic disease as measured by intravascular ultrasound (IVUS).
Enrolled patients were required to be on stable background lipid-lowering therapy and to have a LDL-C ≥80 mg/dL (2.07 mmol/L) or LDL-C ≥60 to <80 mg/dL (1.55 to 2.07 mmol/L) with one major or three minor cardiovascular risk factors. These patients had coronary artery disease and required coronary angiography.
A total of 970 patients were randomized 1:1 into two treatment groups to either receive Repatha 420 mg once monthly or placebo once monthly subcutaneous injections for 76 weeks. IVUS was performed at baseline and at week 78. A total of 27.8% of patients were female, and 93.8% were white. The mean (SD) age was 59.8 (9.2) years. The mean (SD) LDL-C at baseline was 92.6 (27.3) mg/dL (2.4 [0.7] mmol/L).
Repatha reduced percent atheroma volume (PAV) and total atheroma volume (TAV) from baseline to week 78 compared to placebo. Atherosclerosis regression, defined as any reduction in PAV or TAV at week 78, was observed in more patients treated with Repatha than patients treated with placebo.
The results of the study are shown in Table 9 as follows: (See Table 9.)
Click on icon to see table/diagram/image
The treatment difference in LDL-C reduction between Repatha and placebo was 68.7% (95% CI: 64.7%, 72.7%) from baseline to week 78. These reductions were maintained through the end of the study.
Corresponding mean (SD) LDL-C concentrations at week 78 were 29.2 (27.6) mg/dL in the Repatha group.
Based on an ad-hoc analysis, lower LDL-C concentrations achieved during the study were associated with greater atherosclerosis regression, as measured by reduction in PAV.
Effect on coronary atherosclerotic plaque morphology: The effects of Repatha 420 mg once monthly on coronary atherosclerotic plaques as assessed by optical coherence tomography (OCT), were evaluated in a 52-week double-blind, randomised, placebo controlled study including adult patients initiated within 7 days of a non-ST-segment elevation acute coronary syndrome (NSTEACS) on maximally tolerated statin therapy. For the primary endpoint of absolute change in minimum FCT (fibrous cap thickness) in a matched segment of artery from baseline, least squares (LS) mean (95% CI) increased from baseline by 42.7 μm (32.4, 53.1) in the Repatha group and 21.5 μm (10.9, 32.1) in the placebo group, an additional 21.2 μm (4.7, 37.7) compared to placebo (p=0.015; 38% difference (p=0.041)). The reported secondary findings show treatment differences including change in mean minimum FCT (increase 32.5 μm (12.7, 52.4); p=0.016) and absolute change in maximum lipid arc (-26° (-49.6, -2.4); p=0.041).
Effect on LDL-C During Acute Phase of Acute Coronary Syndrome (ACS): EVOPACS was an investigator-sponsored, multicenter, double-blind, randomized, placebo-controlled, 8-week study conducted in Switzerland of Repatha in 308 patients admitted to the hospital within 24 to 72 hours of an ACS event who received concomitant atorvastatin. Repatha 420 mg once monthly significantly reduced LDL-C from baseline to week 8 compared with placebo (p <0.001).
The mean (SD) reduction in calculated LDL-C from baseline at week 8 was 77.1% (15.8%) in the evolocumab group and 35.4% (26.6%) in the placebo group, with a least squares (LS) mean difference (95% CI) of 40.7% (36.2%, 45.2%). Baseline LDL-C values were 3.61 mmol/L (139.5 mg/dL) in the evolocumab group and 3.42 mmol/L (132.2 mg/dL) in the placebo group. LDL-C reductions in this study were consistent with previous studies where evolocumab was added to stable lipid-lowering therapy as demonstrated by on-treatment LDL-C levels at week 8 in this study (reflecting steady-state effect of high-intensity statin in both treatment arms) of 0.79 mmol/L (30.5 mg/dL) and 2.06 mmol/L (79.7 mg/dL) in the evolocumab plus atorvastatin and the placebo plus atorvastatin groups, respectively.
The effects of evolocumab in this patient population were consistent with those observed in previous studies in the evolocumab clinical development program and no new safety concerns were noted.
Pharmacokinetics: Evolocumab exhibits non-linear kinetics as a result of binding to PCSK9. Administration of the 140 mg dose in healthy volunteers resulted in a C
max mean of 18.6 μg/mL and AUC
last mean of 188 day·μg/mL. Administration of the 420 mg dose in healthy volunteers resulted in a C
max mean of 59.0 μg/mL and AUC
last mean of 924 day·μg/mL. Following a single 420 mg intravenous dose, the mean systemic clearance was estimated to be 12 mL/hr. An approximate 2- to 3-fold accumulation was observed in trough serum concentrations (C
min 7.21) following 140 mg doses administered subcutaneously every 2 weeks or following 420 mg doses administered subcutaneously monthly (C
min 11.2), and serum trough concentrations approached steady-state by 12 weeks of dosing.
Absorption: Following a single subcutaneous dose of 140 mg or 420 mg evolocumab administered to healthy adults, median peak serum concentrations were attained in 3 to 4 days, and estimated absolute bioavailability was 72%.
Distribution: Following a single 420 mg intravenous dose, the mean steady-state volume of distribution was estimated to be 3.3 L.
Elimination: Two elimination phases were observed for REPATHA. At low concentrations, the elimination is predominately through saturable binding to target (PCSK9), while at higher concentrations the elimination of REPATHA is largely through a non-saturable proteolytic pathway. REPATHA was estimated to have an effective half-life of 11 to 17 days.
Specific Populations: The pharmacokinetics of evolocumab were not affected by age, gender, race, or creatinine clearance across all approved populations [see Renal Impairment under Precautions].
The exposure of evolocumab decreased with increasing body weight. These differences are not clinically meaningful.
Pediatric Patients: The pharmacokinetics of REPATHA were evaluated in 103 pediatric patients aged 10 to 17 years with HeFH [see Use in Children under Precautions and Clinical Studies as previously mentioned]. Following subcutaneous administration of 420 mg REPATHA once monthly, mean trough serum concentrations were 22.4 mcg/mL and 25.8 mcg/mL over the Week 12 and Week 24 time points, respectively. The pharmacokinetics of REPATHA were evaluated in 12 pediatric patients aged 11 to 17 years with HoFH [see Use in Children under Precautions and Clinical Studies as previously mentioned]. Following subcutaneous administration of 420 mg REPATHA once monthly, mean serum trough concentrations were 20.3 mcg/mL and 17.6 mcg/mL at Week 12 and Week 80, respectively.
Renal Impairment: Since monoclonal antibodies are not known to be eliminated via renal pathways, renal function is not expected to impact the pharmacokinetics of evolocumab.
In a clinical trial of 18 patients with either normal renal function (estimated glomerular filtration rate [eGFR] ≥90 mL/min/1.73 m
2, n=6), severe renal impairment (eGFR <30 mL/min/1.73 m
2, n=6), or end-stage renal disease (ESRD) receiving hemodialysis (n=6), exposure to evolocumab after a single 140 mg subcutaneous dose was decreased in patients with severe renal impairment or ESRD receiving hemodialysis. Reductions in PCSK9 levels in patients with severe renal impairment or ESRD receiving hemodialysis was similar to those with normal renal function [see Hepatic Impairment under Precautions].
Hepatic Impairment: Following a single 140 mg subcutaneous dose of evolocumab in patients with mild or moderate hepatic impairment, a 20-30% lower mean C
max and 40-50% lower mean AUC were observed as compared to healthy patients [see Hepatic Impairment under Precautions].
Drug Interaction Studies: An approximately 20% decrease in the C
max and AUC of evolocumab was observed in adult patients co-administered with a high-intensity statin regimen. This difference is not clinically meaningful.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: The carcinogenic potential of evolocumab was evaluated in a lifetime study conducted in the hamster at dose levels of 10, 30, and 100 mg/kg administered every 2 weeks. There were no evolocumab-related tumors at the highest dose at systemic exposures up to 38- and 15-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. The mutagenic potential of evolocumab has not been evaluated; however, monoclonal antibodies are not expected to alter DNA or chromosomes.
There were no adverse effects on fertility (including estrous cycling, sperm analysis, mating performance, and embryonic development) at the highest dose in a fertility and early embryonic developmental toxicology study in hamsters when evolocumab was subcutaneously administered at 10, 30, and 100 mg/kg every 2 weeks. The highest dose tested corresponds to systemic exposures up to 30- and 12-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. In addition, there were no adverse evolocumab-related effects on surrogate markers of fertility (reproductive organ histopathology, menstrual cycling, or sperm parameters) in a 6-month chronic toxicology study in sexually mature monkeys subcutaneously administered evolocumab at 3, 30, and 300 mg/kg once weekly. The highest dose tested corresponds to 744- and 300-fold the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC.
Animal Toxicology and/or Pharmacology: During a 3-month toxicology study of 10 and 100 mg/kg once every 2 weeks evolocumab in combination with 5 mg/kg once daily rosuvastatin in adult monkeys, there were no effects of evolocumab on the humoral immune response to keyhole limpet hemocyanin (KLH) after 1 to 2 months exposure. The highest dose tested corresponds to exposures 54- and 21-fold higher than the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC. Similarly, there were no effects of evolocumab on the humoral immune response to KLH (after 3 to 4 months exposure) in a 6-month study in cynomolgus monkeys at dose levels up to 300 mg/kg once weekly evolocumab corresponding to exposures 744- and 300-fold greater than the recommended human doses of 140 mg every 2 weeks and 420 mg once monthly, respectively, based on plasma AUC.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image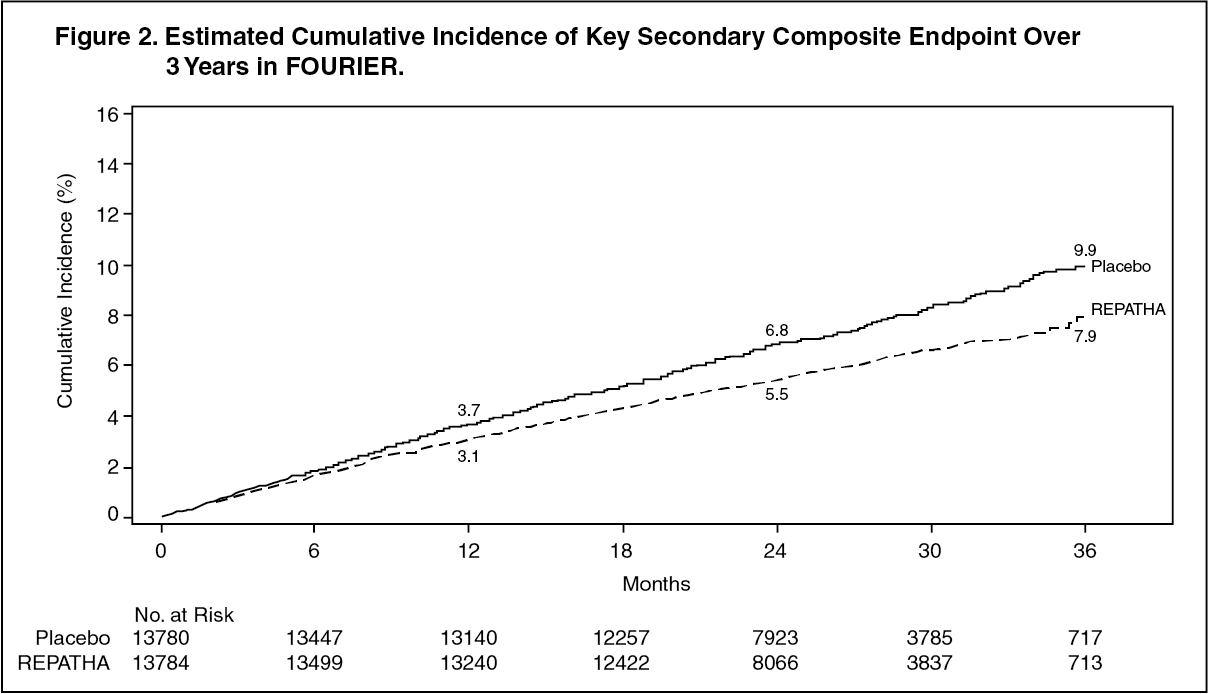 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image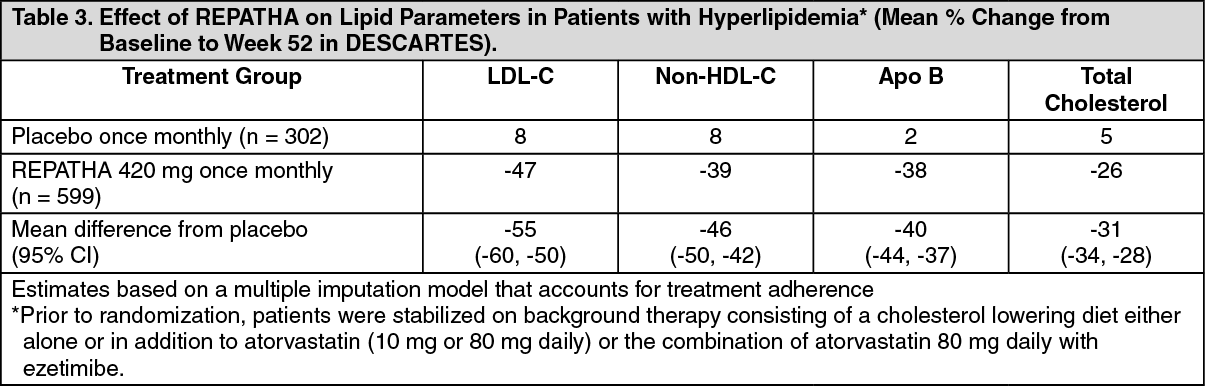 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image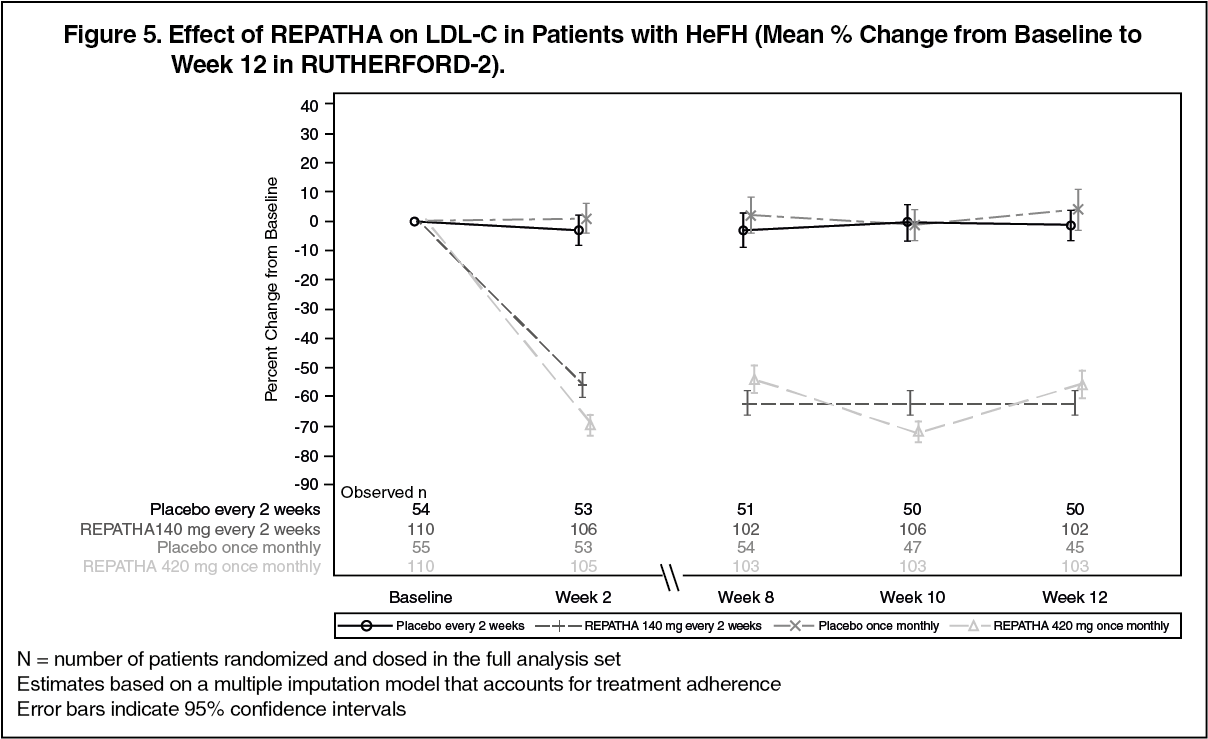 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image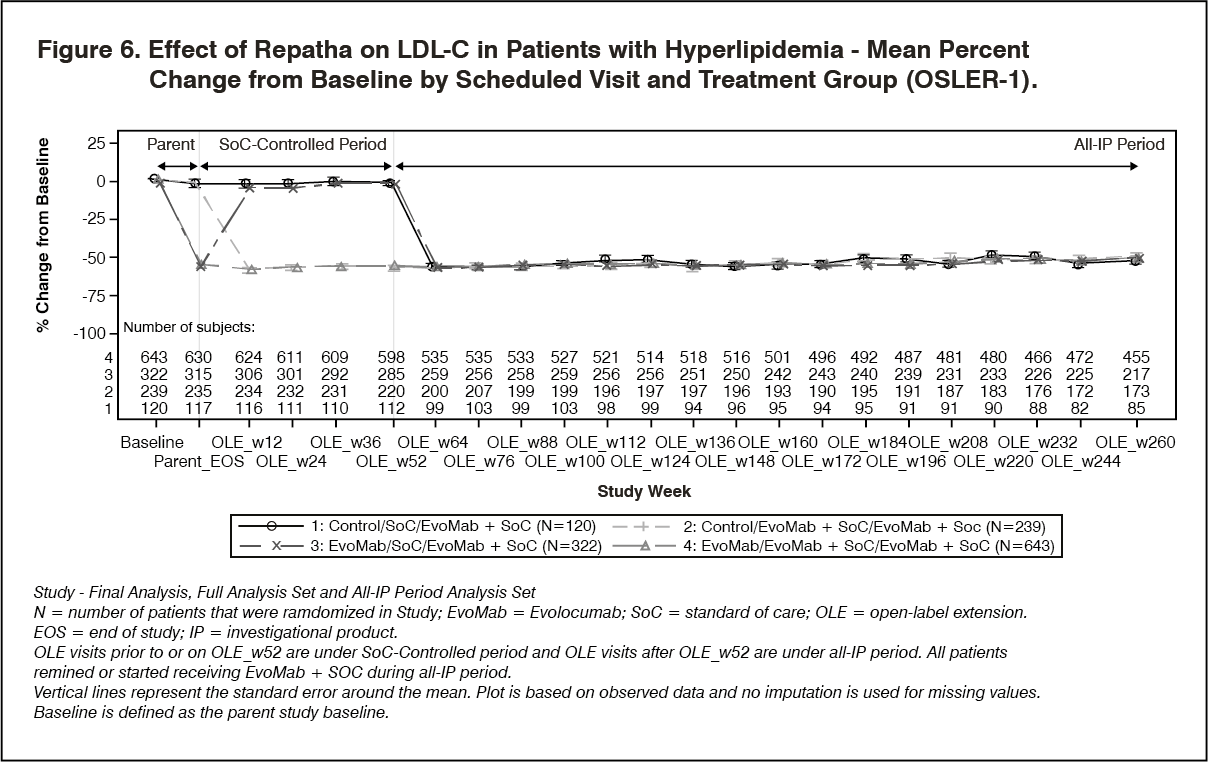 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image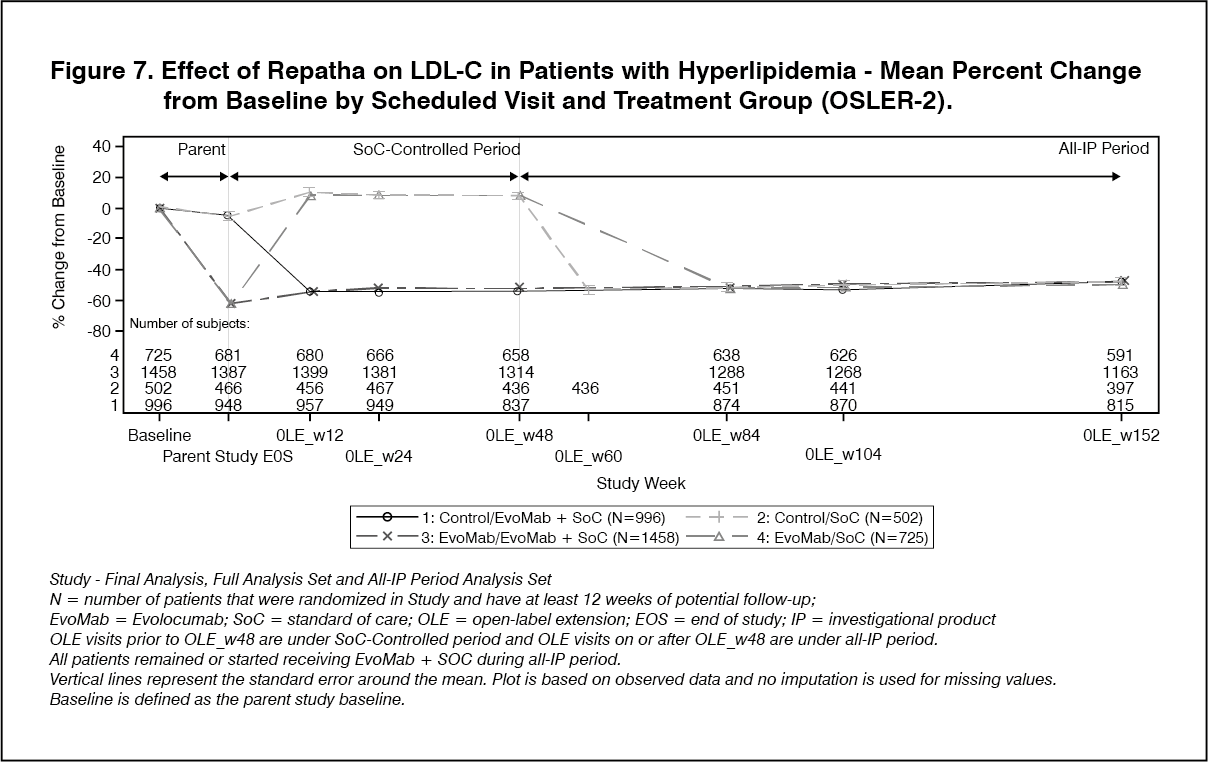 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image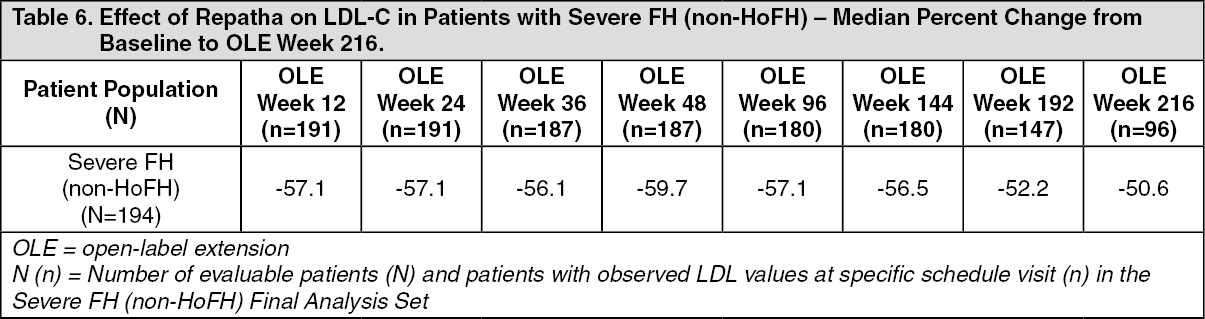 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image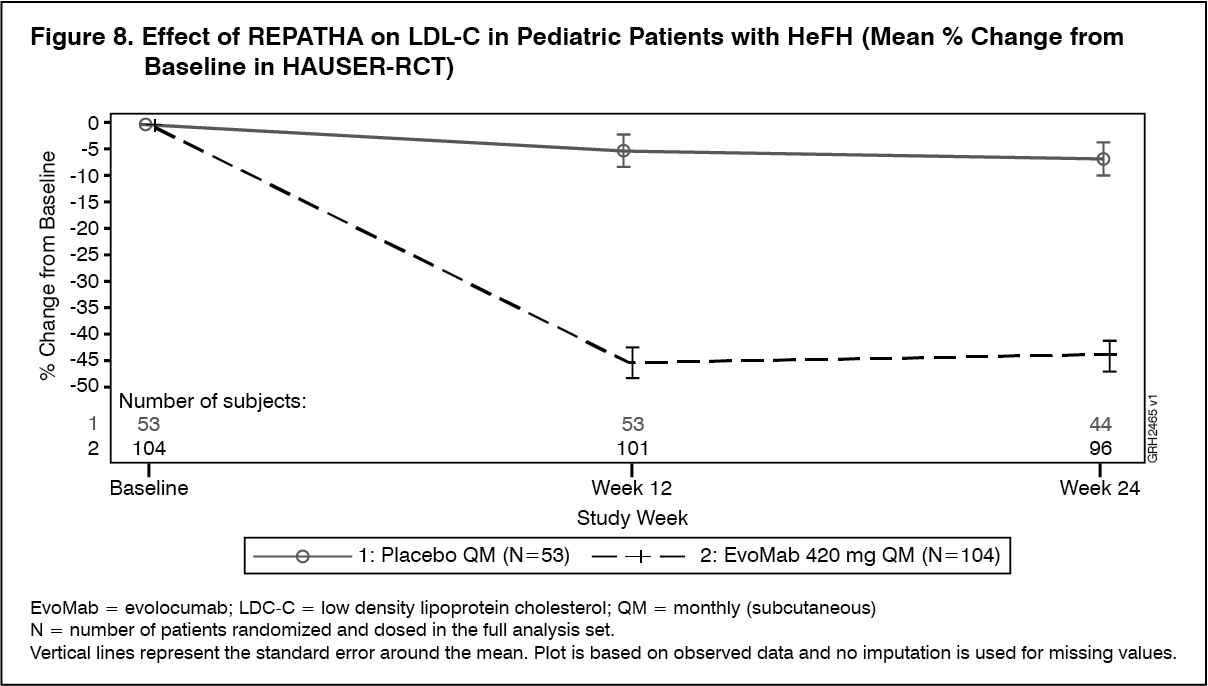 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image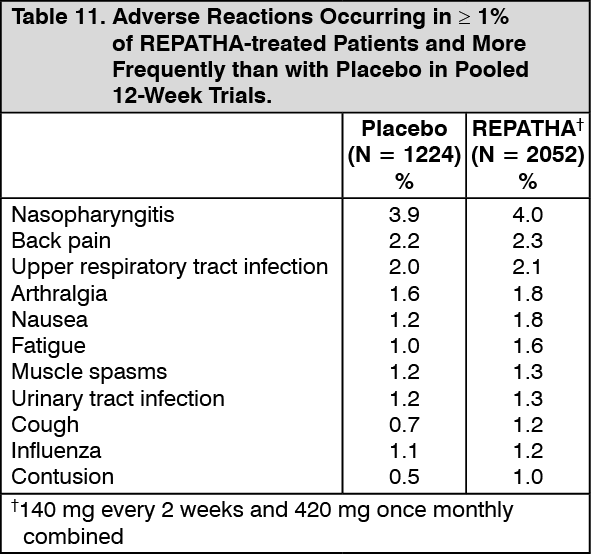 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
