


 Sign Out
Sign Out

Ixazomib induced apoptosis of multiple myeloma cell lines in vitro. Ixazomib demonstrated in vitro cytotoxicity against myeloma cells from patients who had relapsed after multiple prior therapies, including bortezomib, lenalidomide, and dexamethasone. The combination of ixazomib and lenalidomide demonstrated synergistic cytotoxic effects in multiple myeloma cell lines. In vivo, ixazomib demonstrated antitumor activity in a mouse multiple myeloma tumor xenograft model.
Pharmacodynamics: Cardiac Electrophysiology: Ninlaro did not prolong the QTc interval at clinically relevant exposures based on pharmacokinetic-pharmacodynamic analysis of data from 245 patients.
CLINICAL STUDIES: TOURMALINE-MM1: The efficacy and safety of Ninlaro in combination with lenalidomide and dexamethasone was evaluated in a randomized, double-blind, placebo-controlled, multicenter study in patients with relapsed and/or refractory multiple myeloma who had received at least one prior line of therapy.
Patients who were refractory to lenalidomide or proteasome inhibitors were excluded from the study. A total of 722 patients were randomized in a 1:1 ratio to receive either the combination of Ninlaro, lenalidomide and dexamethasone (N=360; Ninlaro regimen) or the combination of placebo, lenalidomide and dexamethasone (N=362; placebo regimen) until disease progression or unacceptable toxicity.
Randomization was stratified according to number of prior lines of therapy (1 versus 2 or 3), myeloma International Staging System (ISS) (stage I or II versus III), and previous therapy with a proteasome inhibitor (exposed or naïve). Twenty three percent (N=166) of the patients had light chain disease and 12% (N=87) of patients had free light chain-measurable only disease.
Thromboprophylaxis was recommended for all patients in both treatment groups according to the lenalidomide prescribing information. Antiemetics were used in 19% of patients in the Ninlaro regimen and 12% of patients in the placebo regimen; antivirals in 64% and 60%, respectively, and antihistamines in 27% and 19%, respectively. These medications were given to patients at the physician's discretion as prophylaxis and/or management of symptoms.
Patients received Ninlaro 4 mg or placebo on Days 1, 8, and 15 plus lenalidomide (25 mg) on Days 1 through 21 and dexamethasone (40 mg) on Days 1, 8, 15, and 22 of a 28-day cycle. Patients with renal impairment received a starting dose of lenalidomide according to its prescribing information. Treatment continued until disease progression or unacceptable toxicities.
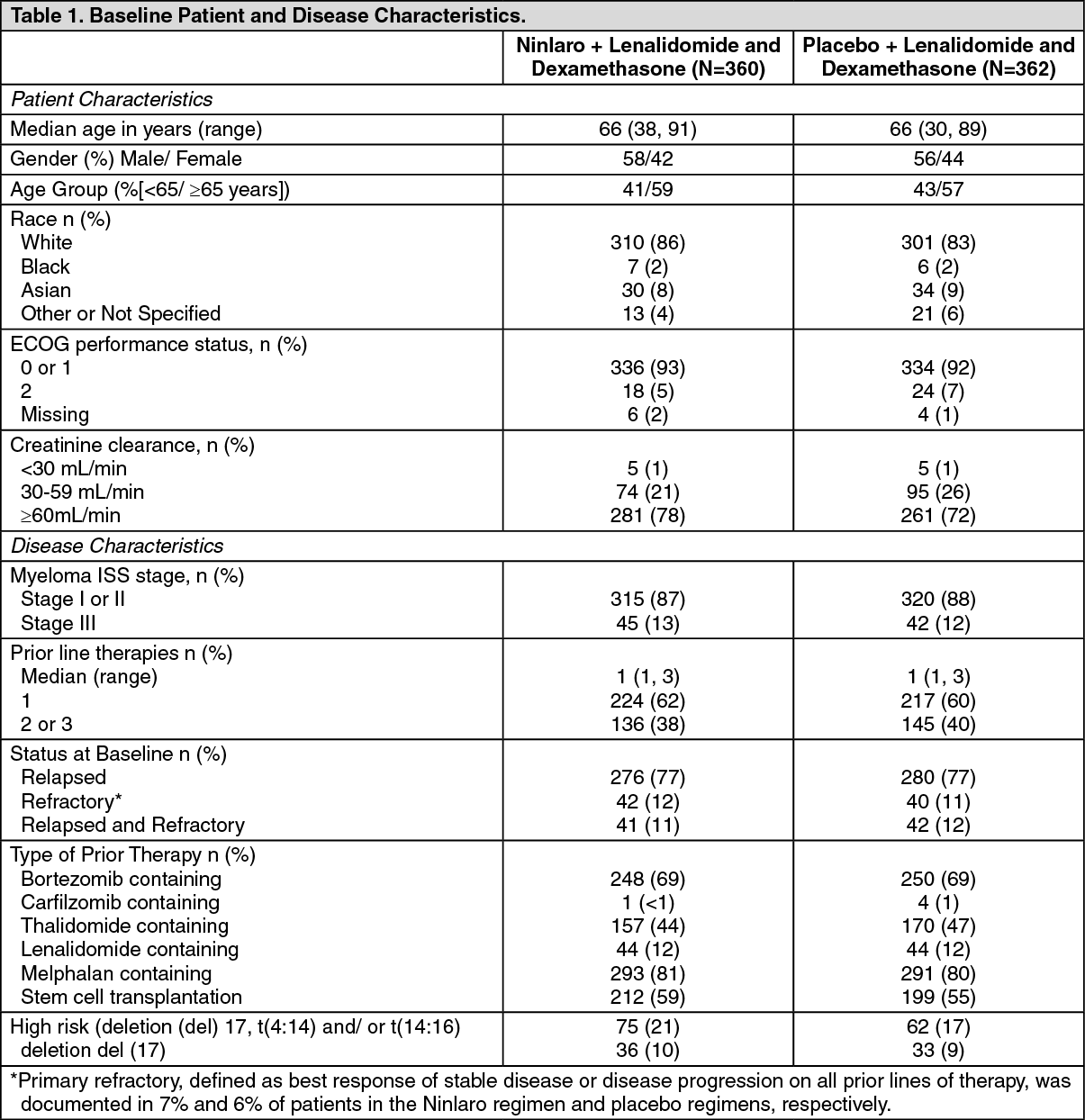
Table 1 summarizes the baseline patient and disease characteristics in the study. The baseline demographics and disease characteristics were balanced and comparable between the study regimens. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe efficacy of Ninlaro was evaluated by progression-free survival (PFS) according to the 2011 International Myeloma Working Group (IMWG) Consensus Uniform Response Criteria as assessed by a blinded independent review committee (IRC) based on central lab results. Response was assessed every four weeks until disease progression.
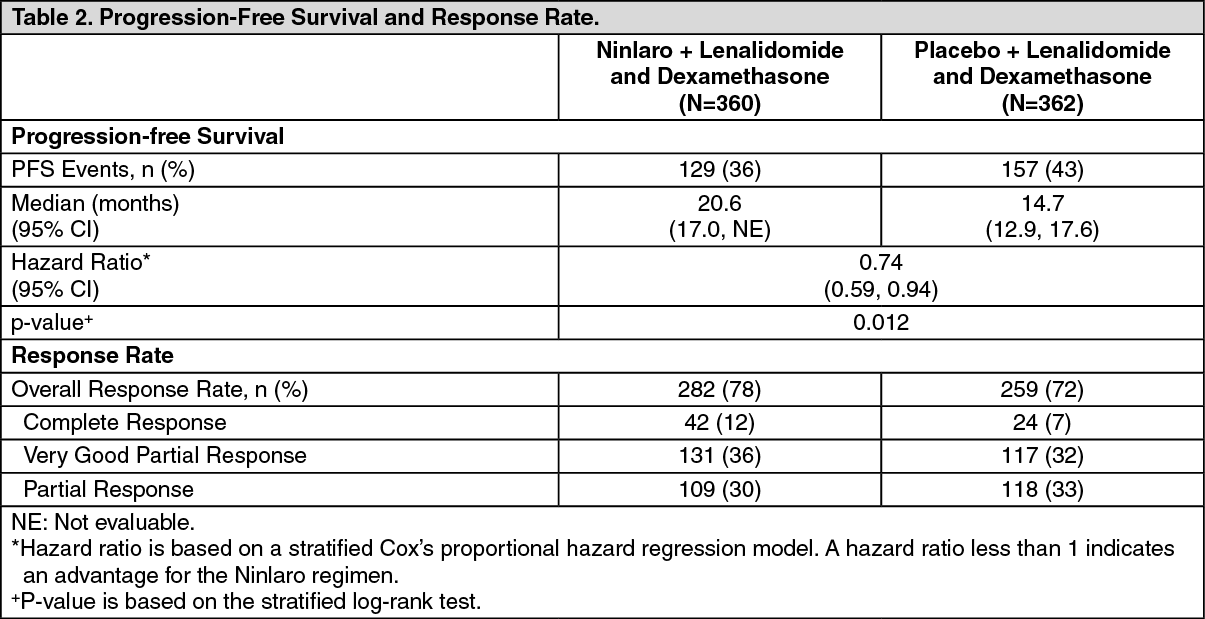
The approval of Ninlaro was based upon a statistically significant improvement in PFS of the Ninlaro regimen compared to the placebo regimen. PFS results are summarized in Table 2 and shown in the figure. (See Table 2.)
 Click on icon to see table/diagram/image
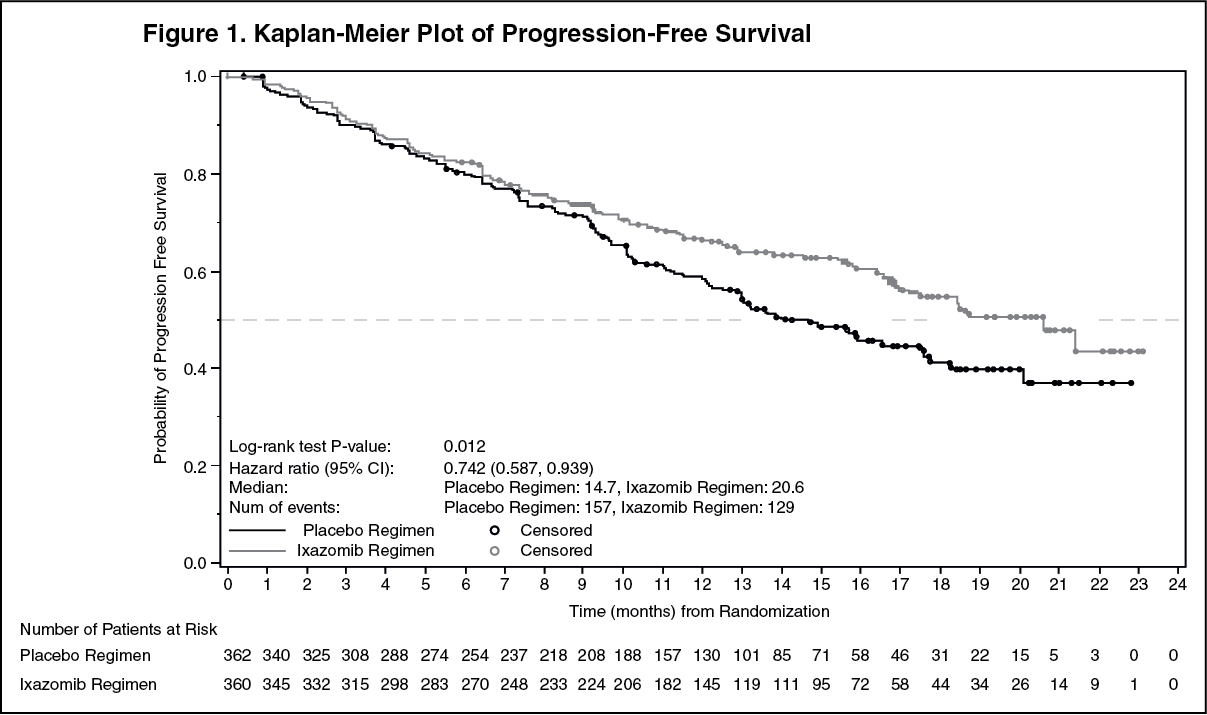
Click on icon to see table/diagram/imageThe median time to response was 1.1 months in the Ninlaro regimen and 1.9 months in the placebo regimen. The median duration of response was 20.5 months in the Ninlaro regimen and 15 months in the placebo regimen for responders in the response evaluable population. (See figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA second, non-inferential, PFS analysis was conducted with a median follow up of 23 months. At this analysis, estimated median PFS was 20 months in the ixazomib regimen and 15.9 months in the placebo regimen (HR=0.82 [95% CI (0.67, 1.0)]) in the ITT population. For patients with one prior therapy, the median PFS was 18.7 months in the ixazomib regimen and 17.6 months in the placebo regimen (HR = 0.99). For patients with 2 or 3 prior therapies, PFS was 22.0 months in the ixazomib regimen and 13.0 months in the placebo regimen (HR = 0.62).
At the final analysis for OS at a median duration of follow up of approximately 85 months, median OS in the ITT population was 53.6 months for patients in the ixazomib regimen and 51.6 months for patients in the placebo regimen (HR = 0.94 [95% CI: 0.78, 1.13; p=0.495]). For patients with one prior therapy, the median OS was 54.3 months in the ixazomib regimen and 58.3 months in the placebo regimen (HR = 1.02 [95% CI: 0.80, 1.29]). For patients with 2 or 3 prior therapies, the median OS was 53.0 months in the ixazomib regimen and 43.0 months in the placebo regimen (HR = 0.85 [95% CI: 0.64, 1.11]).
A randomised, double blind, placebo controlled Phase 3 study was conducted in China (N=115) with a similar study design and eligibility criteria. Many of the patients enrolled in the study had advanced disease with Durie Salmon Stage III (69%) at initial diagnosis and a treatment history of receiving at least 2 prior therapies (60%) and being thalidomide refractory (63%). At the primary analysis (median follow up of 8 months and a median of 6 cycles), the median PFS was 6.7 months in the ixazomib regimen compared to 4 months in the placebo regimen (p value=0.035, HR=0.60). At the final analysis for OS at a median follow up of 19.8 months, OS was improved for patients treated in the ixazomib regimen compared with placebo [p value=0.0014, HR=0.42, 95% CI: 0.242, 0.726]).
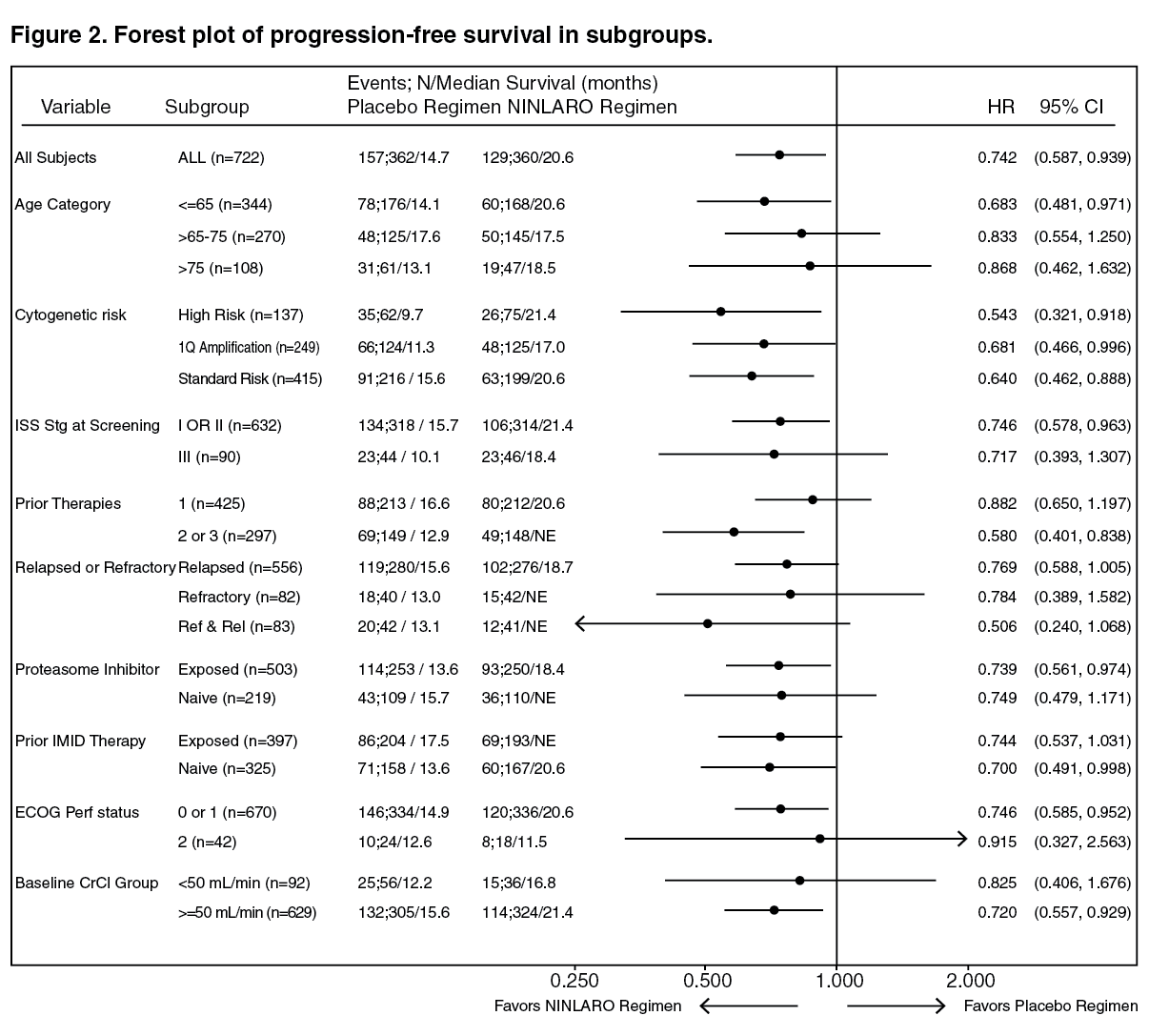
As multiple myeloma is a heterogeneous disease, benefit may vary across subgroups in the Phase 3 study (C16010) (see Figure 2). (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the Phase 3 study (C16010), 10 patients (5 in each treatment regimen) had severe renal impairment at baseline. Of the 5 patients in the ixazomib regimen, one patient had a confirmed partial response and 3 confirmed stable disease (however 2 were unconfirmed partial response and 1 was an unconfirmed very good partial response). Of the 5 patients in the placebo regimen, 2 had a confirmed very good partial response.
Quality of life as assessed by global health scores (EORTC QLQ-C30 and MY-20) was maintained during treatment and was similar in both treatment regimens in the Phase 3 study (C16010).
Pharmacokinetics: Absorption: After oral administration, the median time to achieve peak ixazomib plasma concentrations was one hour. The mean absolute oral bioavailability was 58%, based on population PK analysis. Ixazomib AUC increases in a dose proportional manner over a dose range of 0.2 to 10.6 mg.
A food effect study conducted in patients with a single 4 mg dose of ixazomib showed that a high-fat meal decreased ixazomib AUC by 28% and Cmax by 69% [see Dosage & Administration].
Distribution: Ixazomib is 99% bound to plasma proteins and distributes into red blood cells with a blood-to-plasma ratio of 10. The steady-state volume of distribution is 543 L.
Metabolism: After oral administration of a radiolabeled dose, ixazomib represented 70% of total drug-related material in plasma. Metabolism by multiple CYP enzymes and non-CYP proteins is expected to be the major clearance mechanism for ixazomib. At clinically relevant ixazomib concentrations, in vitro studies using human cDNA-expressed cytochrome P450 isozymes showed that no specific CYP isozyme predominantly contributes to ixazomib metabolism. At higher than clinical concentrations, ixazomib was metabolized by multiple CYP isoforms with estimated relative contributions of 3A4 (42%), 1A2 (26%), 2B6 (16%), 2C8 (6%), 2D6 (5%), 2C19 (5%) and 2C9 (< 1%).
Elimination: Based on a population PK analysis, systemic clearance was approximately 1.9 L/hr with inter-individual variability of 44%. The terminal half-life (t1/2) of ixazomib was 9.5 days. Following weekly oral dosing, the accumulation ratio was determined to be 2-fold.
Excretion: After administration of a single oral dose of 14C-ixazomib to 5 patients with advanced cancer, 62% of the administered radioactivity was excreted in urine and 22% in the feces. Unchanged ixazomib accounted for < 3.5% of the administered dose recovered in urine.
Special Populations: Age, Sex, Race: There was no clinically meaningful effect of age (range 23-91 years), sex, body surface area (range 1.2-2.7 m2), or race on the clearance of ixazomib based on population PK analysis.
Hepatic Impairment: The PK of ixazomib was similar in patients with normal hepatic function and in patients with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1-1.5 x ULN and any AST) based on population PK analysis.
The PK of ixazomib was characterized in patients with normal hepatic function at 4 mg (N=12), moderate hepatic impairment at 2.3 mg (total bilirubin > 1.5-3 x ULN, N=13) or severe hepatic impairment at 1.5 mg (total bilirubin > 3 x ULN, N=18). Dose-normalized mean AUC was 20% higher in patients with moderate or severe hepatic impairment as compared to patients with normal hepatic function [see Dosage & Administration].
Renal Impairment: The PK of ixazomib was similar in patients with normal renal function and in patients with mild or moderate renal impairment (creatinine clearance ≥ 30 mL/min) based on population PK analysis.
The PK of ixazomib was characterized at a dose of 3 mg in patients with normal renal function (creatinine clearance ≥ 90 mL/min, N=18), severe renal impairment (creatinine clearance < 30 mL/min, N=14), or ESRD requiring dialysis (N=6). Mean AUC was 39% higher in patients with severe renal impairment or ESRD requiring dialysis as compared to patients with normal renal function. Pre- and post-dialyzer concentrations of ixazomib measured during the hemodialysis session were similar, suggesting that ixazomib is not dialyzable [see Dosage & Administration].
Drug Interactions: Effect of Other Drugs on Ninlaro: Strong CYP3A Inducers: Co-administration of Ninlaro with rifampin decreased ixazomib Cmax by 54% and AUC by 74% [see Interactions].
Strong CYP3A Inhibitors: Co-administration of Ninlaro with clarithromycin did not result in a clinically meaningful change in the systemic exposure of ixazomib.
Strong CYP1A2 Inhibitors: Co-administration of Ninlaro with strong CYP1A2 inhibitors did not result in a clinically meaningful change in the systemic exposure of ixazomib based on a population PK analysis.
Effect of Ninlaro on Other Drugs: Ixazomib is neither a reversible nor a time-dependent inhibitor of CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4/5. Ixazomib did not induce CYP1A2, CYP2B6, and CYP3A4/5 activity or corresponding immune-reactive protein levels. Ninlaro is not expected to produce drug-drug interactions via CYP inhibition or induction.
Transporter-Based Interactions: Ixazomib is a low affinity substrate of P-gp. Ixazomib is not a substrate of BCRP, MRP2 or hepatic OATPs. Ixazomib is not an inhibitor of P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1, or MATE2-K. Ninlaro is not expected to cause transporter-mediated drug-drug interactions.
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Ixazomib was not mutagenic in a bacterial reverse mutation assay (Ames assay). Ixazomib was considered positive in an in vitro clastogenicity test in human peripheral blood lymphocytes.
However, in vivo, ixazomib was not clastogenic in a bone marrow micronucleus assay in mice and was negative in an in vivo comet assay in mice, as assessed in the stomach and liver. No carcinogenicity studies have been performed with ixazomib.
Developmental toxicity studies in rats and rabbits did not show direct embryo-fetal toxicity below maternally toxic doses of ixazomib. Studies of fertility and early embryonic development and pre- and post-natal toxicology were not conducted with ixazomib, but evaluation of reproductive tissues was conducted in the general toxicity studies. There were no effects due to ixazomib treatment on male or female reproductive organs in studies up to 6-months duration in rats and up to 9-months duration in dogs.