


 Sign Out
Sign Out



SGLT2 lnhibition: Luseogliflozin selectively inhibited the glucose uptake mediated by human SGLT2 (SGLT2-overexpressing cells) (Ki value: 1.1 nmol/L) (in vitro).
Urinary Glucose Excretion: In obese type 2 diabetes models (Zucker Fatty rats and db/db mice), single oral administration increased urinary glucose excretion (8 or 24 hours after the administration). In a non-obese type 2 diabetes model (GK rats), dietary administration for 20 weeks increased urinary glucose excretion (24 hours after the administration).
To patients with type 2 diabetes mellitus, 2.5 mg or 5 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 7 days. Luseogliflozin increased urinary glucose excretion up to 24 hours after the administration compared with placebo.
Hypoglycemic Action: In an obese type 2 diabetes model (Zucker Fatty rats), single oral administration inhibited the increase in plasma glucose after glucose loading. In another obese type 2 diabetes model (db/db mice), once-daily repeat oral administration for 4 weeks decreased the change in glycated hemoglobin from baseline. In a non-obese type 2 diabetes model (GK rats), dietary administration for 20 weeks decreased g!ycated hemoglobin.
To patients with type 2 diabetes mellitus, 2.5 mg or 5 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 7 days. Luseogliflozin improved plasma glucose AUC 4 hours after breakfast, lunch, or dinner and fasting plasma glucose compared with placebo.
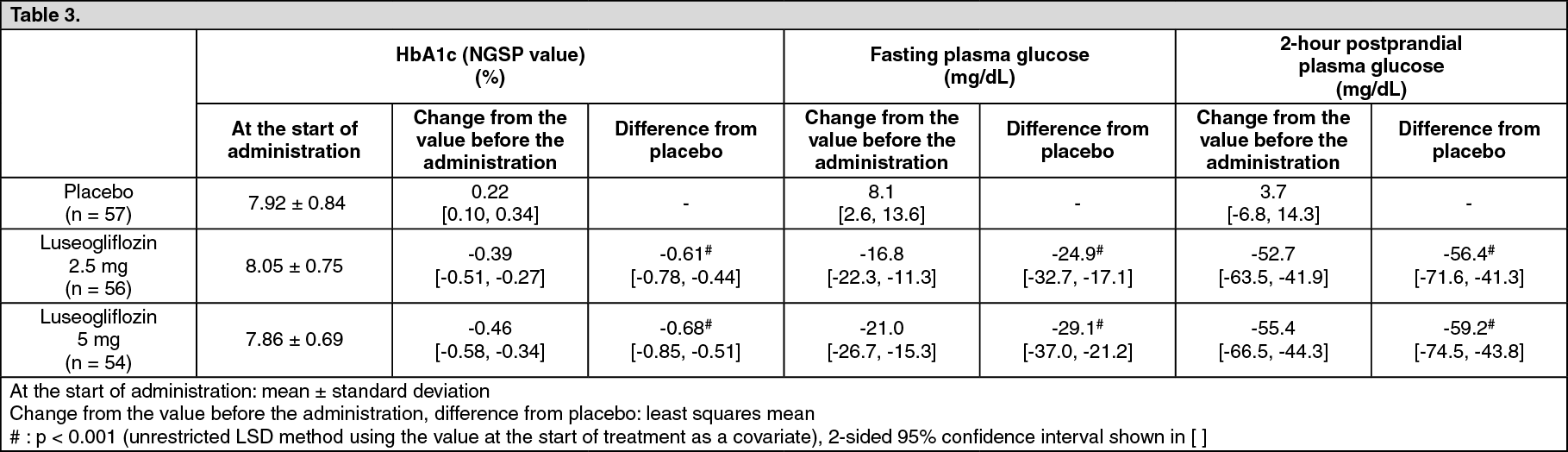
Clinical Studies: Monotherapy: Double-blind placebo-controlled study (dose-finding study): To patients with type 2 diabetes mellitus whose plasma glucose is insufficiently controlled by diet and exercise therapies (280 subjects), 1 mg, 2.5 mg, 5 mg, or 10 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 12 weeks. When changes compared with the value before the administration were examined, luseogliflozin significantly lowered HbA1c (NGSP value) compared with placebo. (See Table 3.)
 Click on icon to see table/diagram/image
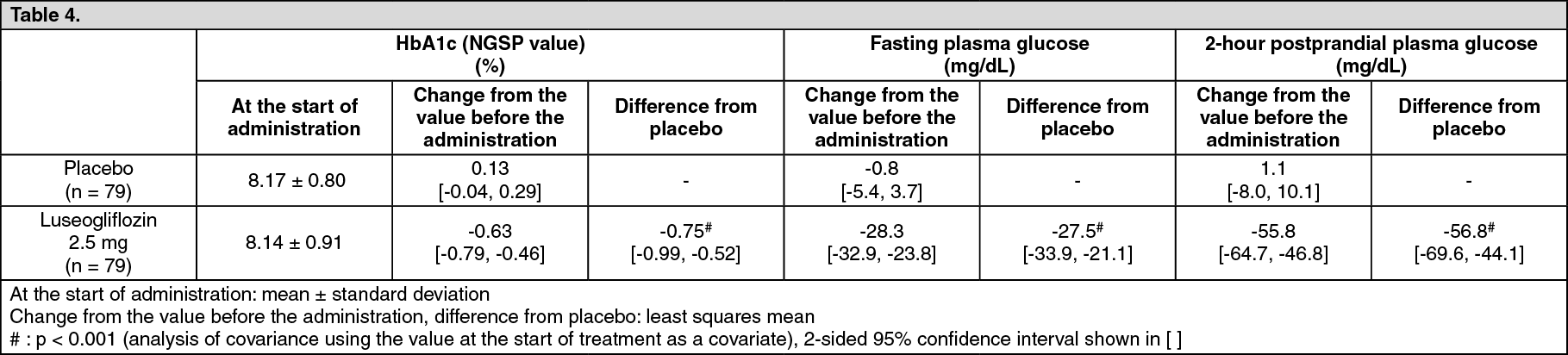
Click on icon to see table/diagram/imageDouble-blind placebo-controlled study (confirmatory study): To patients with type 2 diabetes mellitus whose plasma glucose is insufficiently controlled by diet and exercise therapies (158 subjects), 2.5 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 24 weeks. When changes compared with the value before the administration were examined, luseogliflozin significantly lowered HbA1c (NGSP value) compared with placebo. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLong-term studies: To patients with type 2 diabetes mellitus whose plasma glucose is insufficiently controlled by diet and exercise therapies (299 subjects), 2.5 mg or 5 mg (when the dose was increased) of luseogliflozin was orally administered once daily before breakfast for 52 weeks (HbA1c (NGSP value) at the start of administration: 7.67% ± 0.66%). Luseogliflozin lowered HbA1c (NGSP value) starting from early in the administration and the change in HbA1c (NGSP value) at Week 52 from the start of the administration (mean (2-sided 95% confidence interval)) was -0.50 (-0.6, -0.4)%. Stable glycemic control was achieved throughout the 52 weeks. The incidence of the adverse drug reaction of hypoglycemia was 1.3% (4/299 subjects).
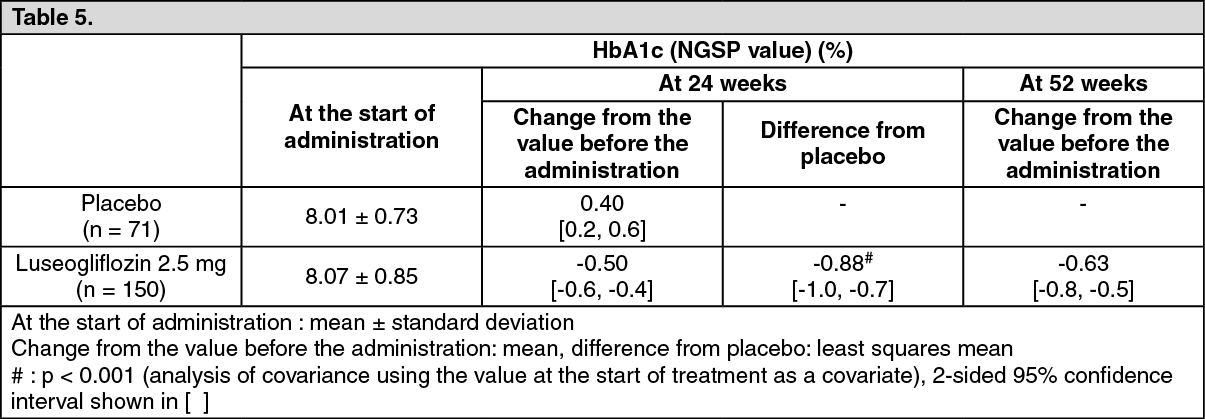
Add-on combination therapy: Long-term study of luseogliflozin in add-on combination with sulfonylurea: To patients with type 2 diabetes mellitus whose plasma control was insufficiently controlled by diet and exercise therapies and monotherapy with glimepiride (150 subjects), 2.5 mg or 5 mg (when the dose was increased) of luseogliflozin was orally administered once daily before breakfast for 52 weeks. Luseogliflozin lowered HbA1c (NGSP value) starting from early in the administration. Stable glycemic control was achieved throughout the 52 weeks in combined use with glimepiride. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe incidence of the adverse drug reaction of hypoglycemia was 8.7% (13/150 subjects) in combined use with glimepiride.
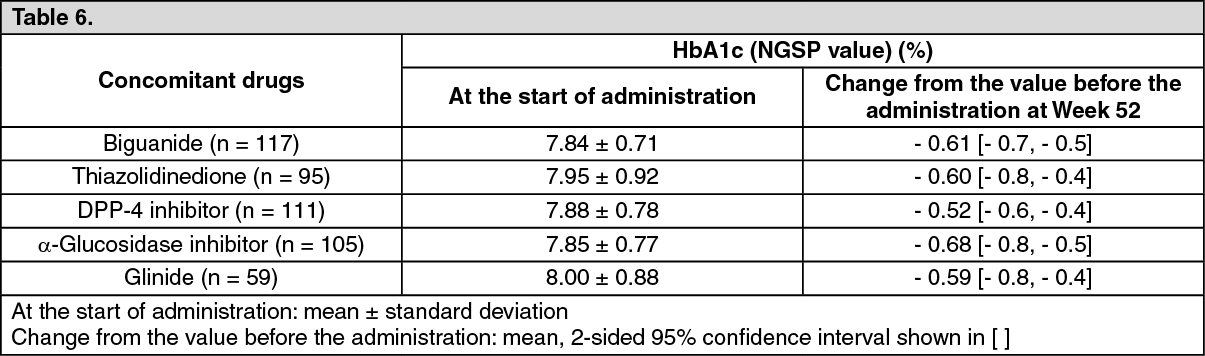
Long-term study of luseogliflozin in add-on combination with oral hypogylcemic drugs: To patients with type 2 diabetes mellitus whose plasma control was insufficiently controlled by diet and exercise therapies and monotherapy with oral hypoglycemic drugs (biguanide (117 subjects), thiazolidinedione (95 subjects), DPP-4 inhibitor (111 subjects), α-glucosidase inhibitor (105 subjects), glinide (59 subjects)), 2.5 mg or 5 mg (when the dose was increased) of luseogliflozin was orally administered once daily before breakfast for 52 weeks. Luseogliflozin lowered HbA1c (NGSP value) starting from early in the administration. Stable glycemic control was achieved throughout the 52 weeks in combined use with any of the oral hypoglycemic drugs examined. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe incidence of the adverse drug reaction of hypoglycemia was 2.6% (3/117 subjects) in combined use with biguanide, 2.1% (2/95 subjects) in combined use with thiazolidinedione, 0.9% (1/111 subjects) in combined use with DPP-4 inhibitor and 1.7% (1/59 subjects) in combined use with glinide. No hypoglycemia was observed in combined use with α-glucosidase inhibitor.
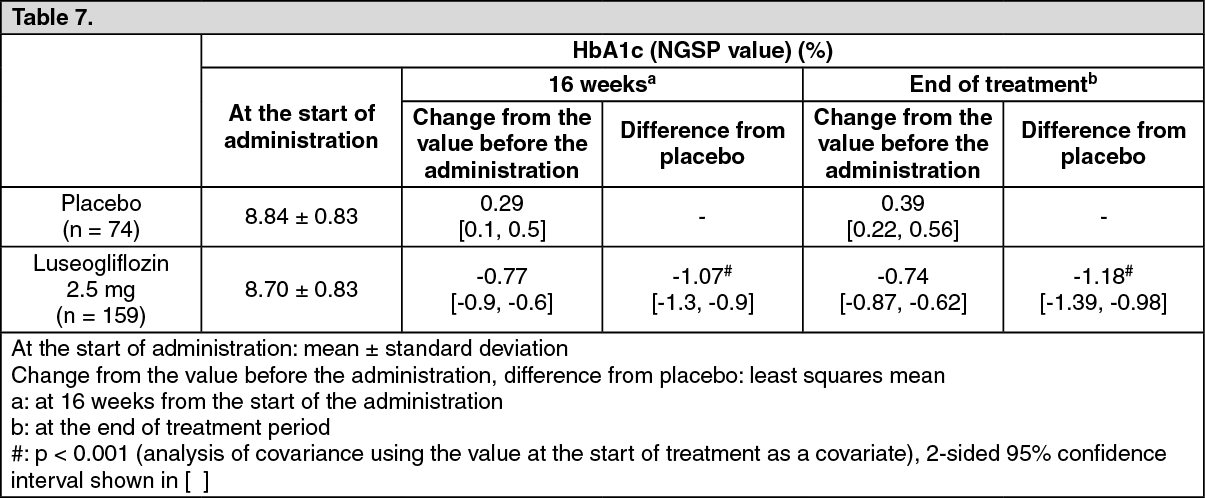
Long-term use of luseogliflozin in add-on combination with insulin preparations: To patients with type 2 diabetes mellitus whose plasma control was insufficiently controlled by diet and exercise therapies and insulin preparation (233 subjects), 2.5 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 16 weeks. The results were as the following table. The incidence of the adverse drug reaction of hypoglycemia were 10.7% in the placebo group (8/74 subjects) and 18.9% in the luseogliflozin group (30/159 subjects). (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn patients who were administered with luseogliflozin continuously for 52 weeks as the result of assignment in the luseogliflozin group in the double-blind treatment period for 16 weeks and proceeding to the open-label treatment period for 36 weeks, change in HbA1c (NGSP value) (mean [2-sided 95% confidence interval]) was -1.00% (-1.1%, -0.9%) from the start of the administration. The incidence of the adverse drug reaction of hypoglycemia was 29.6% (47/159 subjects) in the 52 weeks administration group.
Efficacy in Patients with Renal lmpairment: When 2.5 mg of luseogliflozin or placebo was orally administered once daily before breakfast for 24 weeks in type 2 diabetic patients with moderate renal impairment (eGFR, 30 ml/min/1.73 m2 or higher, 59 mL/min/1.73 m2 or lower) (145 subjects), change in HbA1c was as follows. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen, in addition to the above administration, 2.5 mg or 5 mg (when the dose was increased) of luseogliflozin was administered once daily for 28 weeks (52 weeks in total) (95 subjects) (HbA1c (NGSP value) at the start of the administration: 7.72% ± 0.68%), change from the start of the administration in HbA1c (NGSP value) (mean (2-sided 95% confidence interval)) was - 0.30 (- 0.4, - 0.2)%.
Pharmacokinetics: Plasma Concentrations: Single administration: In single oral administration of luseogliflozin in fasting condition at a dose of 2.5 mg in healthy male adults, time-course change in plasma concentration and pharmacokinetic parameters of luseogliflozin and its active metabolite, M2, were as follows. (See figure and Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
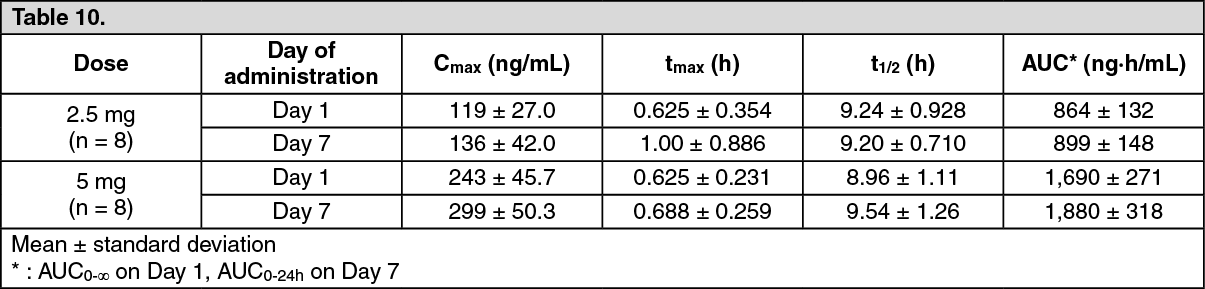
Click on icon to see table/diagram/imageRepeat administration: In 7-day once-daily repeat oral administration of luseogliflozin at a dose of 2.5 mg or 5 mg in patients with type 2 diabetes mellitus, pharmacokinetic parameters of luseogliflozin were as follows. The molar ratio of the active metabolite, M2, to luseogliflozin calculated from the AUC0-24h on Day 7 of the administration was 14.0% and 14.8% at doses of 2.5 mg and 5 mg, respectively. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffects of food intake: When luseogliflozin was orally administered in fasting condition, 5 minutes before breakfast (before meal) or 30 minutes after breakfast (after meal) in a single dose of 2.5 mg in healthy male adults (9 subjects), the geometric mean ratios of Cmax and AUC0-72h and their 90% confidence intervals were 0.790 [0.670, 0.933] and 0.986 [0.958, 1.01] for after meal/before meal, 0.922 [0.781, 1.09] and 0.980 [0.953, 1.01] for fasting/before meal, 0.857 [0.726, 1.01] and 1.01 [0.977, 1.04] for after meal/fasting, and 1.08 [0.919, 1.28] and 1.02 [0.991, 1.05] for before meal/fasting.
Protein Binding: The protein binding in human plasma was 96.0% to 96.3% at concentrations of 50 to 5,000 ng/ml (in vitro, ultracentrifugation).
Metabolism: As the main metabolites in plasma and urine in oral administration of luseogliflozin in healthy male adults, O-deethyl form (M2), carboxyl form generated by oxidation after hydroxylation of the terminal carbon of ethyl group (M17), the glucuronide of luseogliflozin (M8), and the glucuronide of M2 (M12) were observed. M2 is the active metabolite which inhibits SGLT2. The 50% inhibitory concentration (IC50 value) of luseogliflozin and M2 for glucose uptake mediated by human SGLT2 (SGLT2-overexpressing cells) were 2.26 and 4.01 nmol/L, respectively (in vitro).
Metabolism of luseogliflozin was shown to mainly involve CYP3A4/5, 4A11, 4F2, 4F3B and UGT1A1 (in vitro).
Luseogliflozin showed weak inhibitory effect on CYP2C19 (IC50 value: 58.3 μmol/L), while it did not show any inhibitory effect on CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, or 3A4 (IC50 > 100 μmol/L) (in vitro). Luseogliflozin was shown not to induce CYP1A2 or 2B6, but to weakly induce CYP3A4 (In vitro). In a study in patients with type 2 diabetes mellitus using urinary 6β-hydroxycortisol concentration as a marker, luseogliflozin did not induce CYP3A4 (data in non-Japanese subjects).
Excretion: In single oral administration of luseogliflozin in fasting condition at a dose of 2.5 mg in healthy male adults (9 subjects), urinary excretion of luseogliflozin up to 72 hours after the administration was 4.47% (mean).
Luseogliflozin was shown to be a substrate of P-glycoprotein (P-gp), but not to be a substrate of breast cancer resistance protein (BCRP), organic anion transporting polypeptides (OATP1B1, OATP1B3), organic anion transporters (OAT1, OAT3) or organic cation transporter (OCT2). Luseogliflozin showed weak inhibitory effect on OATP1B3 (IC50 value: 93.1 μmol/L), while it did not show any inhibitory effect on P-gp, BCRP, OATP1B1, OAT1, OAT3, or OCT2 (IC50 > 100 μmol/L) (in vitro).
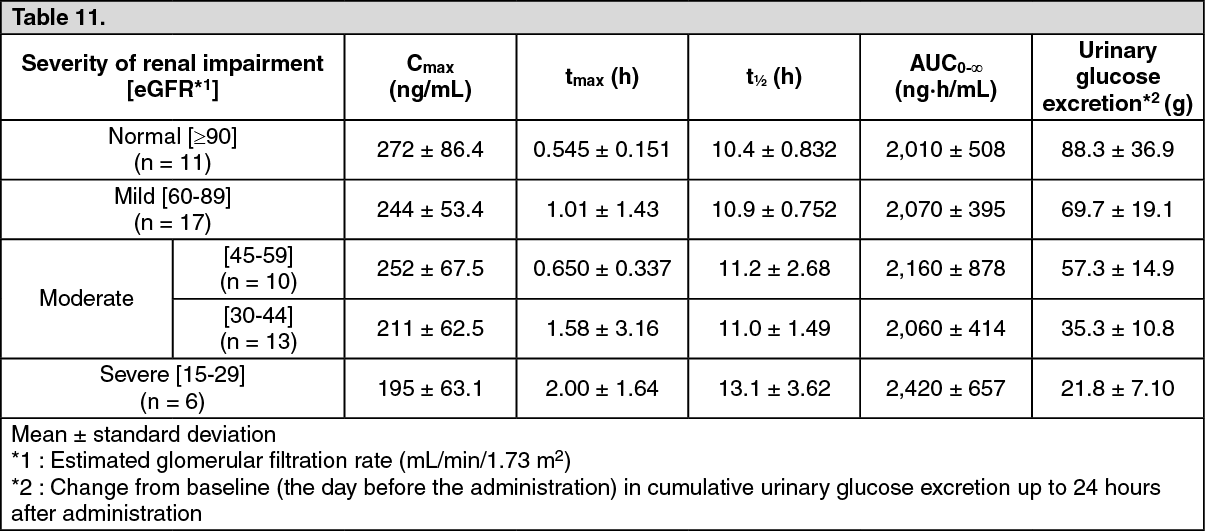
Patients with Renal Impairment: In single oral administration of luseogliflozin at a dose of 5 mg in type 2 diabetic subjects with renal impairment and type 2 diabetic patients with normal renal function, Cmax showed a tendency toward decrease with decline of renal function. (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with Hepatic Impairment: In single oral administration of luseogliflozin at a dose of 5 mg in subjects with hepatic impairment that was up to moderate in severity and subjects with normal liver function, Cmax was 23% lower in the subjects with moderate hepatic impairment than in the subjects with normal liver function. (See Table 12.)
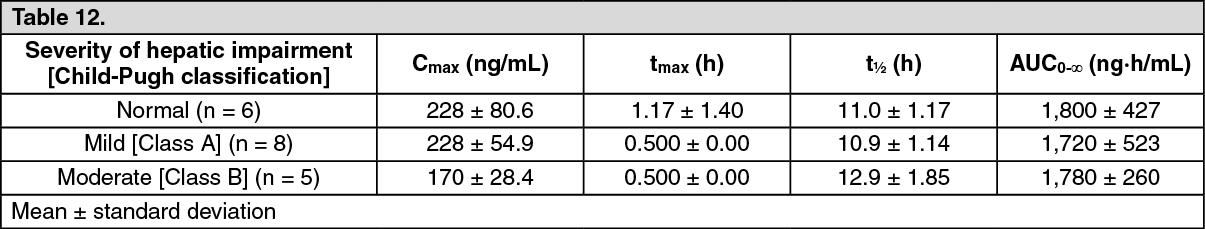 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageElderly Patients: In single oral administration of luseogliflozin at a dose of 5 mg in elderly subjects (24 men and women aged 65 years or older), Cmax, and AUC0-∞ (mean ± standard deviation) were 256 ± 63.6 ng/ml and 2,050 ± 307 ng·h/ml, respectively. In single oral administration of luseogliflozin at a dose of 5 mg in healthy male adults aged between 20 and 40 years (8 subjects) in another study, Cmax, and AUC0-∞ were 205 ± 53.5 ng/ml and 1,930 ± 290 ng·h/ml, respectively.
Drug lnteractions: In combined use of luseogliflozin and various drugs in healthy male adults, the effects of combined use on pharmacokinetic parameters were as follows. (See Table 13.)
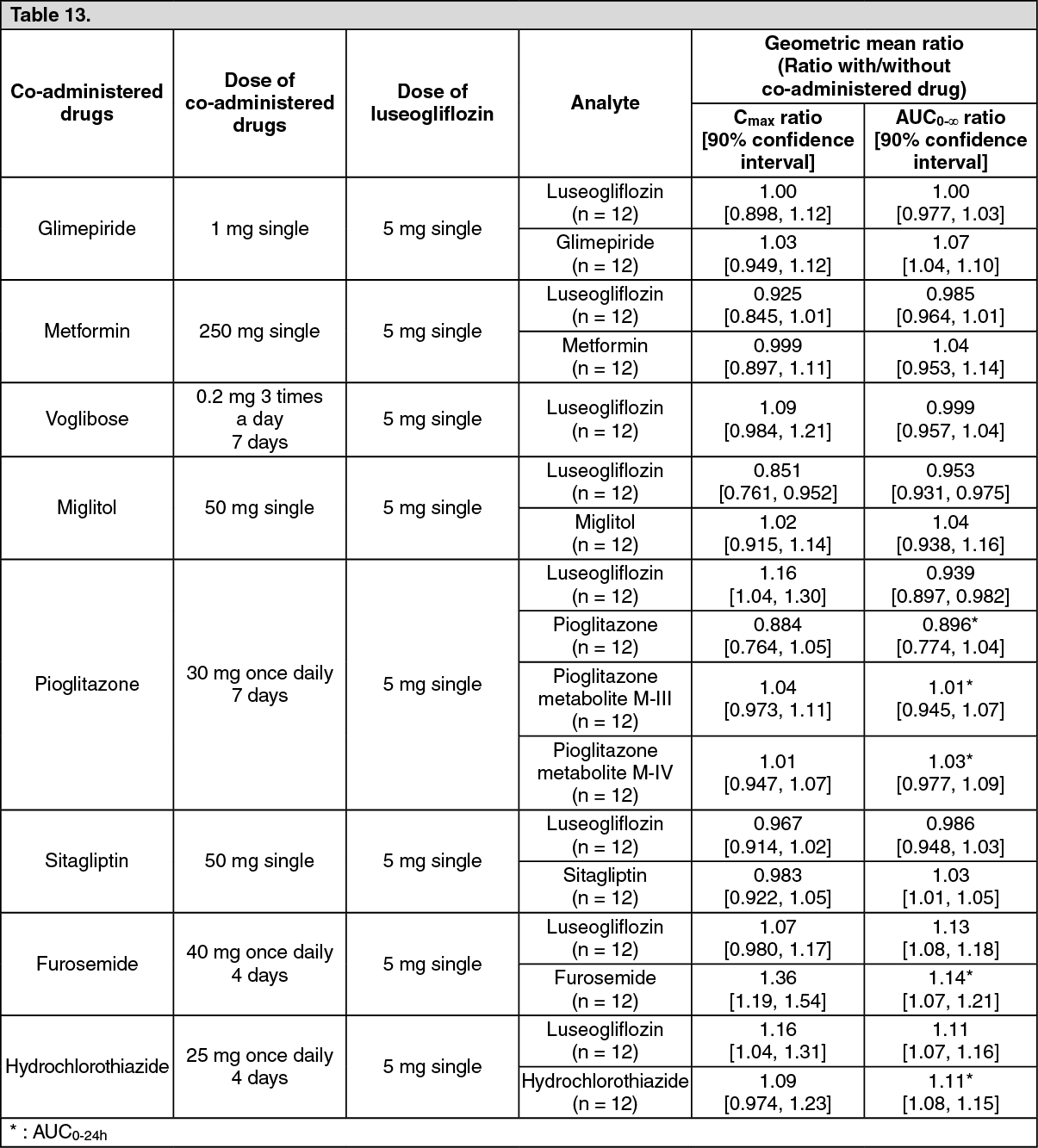 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageToxicology: Preclinical Safety Data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, fertility and early embryonic development, and pre- and postnatal development.
In the carcinogenicity study in mice, no neoplastic change related to luseogliflozin was observed at the highest dose of 100 mg/kg.The AUC0-24h values of luseogliflozin at this dose were 21 to 48 times higher than the AUCτ value in humans. On the other hand,in rats, the numbers of male animals with pheochromocytoma in the adrenals, those with Leydig cell tumor (benign) in the testes, and those with hemangioma/hemangiosarcoma in the mesenteric lymph nodes increased at the highest dose of 100 mg/kg. The AUC0-24h values of luseogliflozin at 20 mg/kg for males and 100 mg/kg for females, at which no neoplastic change was indicated, were 4.2 times higher in males and 40 times higher in females than AUCτ value in humans.
The increased number of rats with pheochromocytoma in the adrenals was probably explained by a change in calcium homeostasis due to persistent SGLT1 inhibition (increased calcium absorption) and increased food consumption (increased calcium consumption). Pheochromocytoma in the adrenals caused through this mechanism of action tends to occur in rats and is poorly extrapolated into humans. In addition, no effects of luseogliflozin on calcium have been reported in humans. It was,therefore, considered very unlikely that luseogliflozin would induce pheochromocytoma in the adrenals in humans.
Leydig cell tumor in the testes was likely caused by increased luteinizing hormone levels due to decreased testosterone levels resulting from long-term repeated administration of luseogliflozin. However, this tumor caused through the above mechanism of action is specific to rats and is poorly extrapolated into humans. It was, therefore, considered very unlikely that luseogliflozin would lead to the occurrence of Leydig cell tumor in humans.
Hemangioma/hemangiosarcoma in the mesenteric lymph nodes was likely caused by the following. The testing facility of the rat carcinogenicity study was prone to develop these tumors in the mesenteric lymph nodes. In addition, it was also suspected that these tumors occurred due to secondary factors, including local ischemia, caused by malnutrition and stress, such as decreased body weight and increased urinary glucose excretion. It was, however, considered very unlikely that luseogliflozin would induce hemangioma/hemangiosarcoma in humans.
In the embryo-fetal development study in rats, low body weight, skeletal variations, delayed ossification, and membranous ventricular septum defect at 150 mg/kg or 500 mg/kg were changes secondary to malnutrition or exacerbation of general condition of dams due to treatment with luseogliflozin. The AUC0-24h value of luseogliflozin at 50 mg/kg, at which no teratogenicity was indicated, was 15 times higher than the AUCτ value in humans.