Strictly follow the recommended dosage unless directed otherwise by the physician. All doses of irinotecan should be administered as an intravenous infusion over 30 to 90 minutes.
Single-agent dosage schedules: Single-agent dosage schedules have been extensively studied for metastatic colorectal cancer.
Starting dose: Weekly Dosage Schedule: The recommended single-agent starting dose of irinotecan is 125 mg/m
2. A lower starting dose may be considered (e.g., 100 mg/m
2) for patients with any of the following conditions: prior extensive radiotherapy, performance status of 2, increased bilirubin levels, or gastric cancer. Treatment should be given in repeated 6-week cycles, comprising weekly treatment for 4 weeks, followed by a 2-week rest.
Once-Every-2-Week Dosage Schedule: The usual recommended starting dose of irinotecan is 250 mg/m
2 every 2 weeks by intravenous infusion. A lower starting dose may be considered (e.g., 200 mg/m
2) for patients with any of the following conditions: age 65 years and older, prior extensive radiotherapy, performance status of 2, increased bilirubin levels, or gastric cancer.
Once-Every-3-Week Dosage Schedule: The usual recommended starting dose of irinotecan for the once-every-3-week dosage schedule is 350 mg/m
2. A lower starting dose may be considered (e.g., 300 mg/m
2) for patients with any of the following conditions: age 65 years and older, prior extensive radiotherapy, performance status of 2, increased bilirubin levels, or gastric cancer.
Special populations: Elderly: The dose should be chosen carefully in this population due to their greater frequency of decreased biological functions. This population should require more intensive surveillance (See Pharmacology: Pharmacokinetics under Actions).
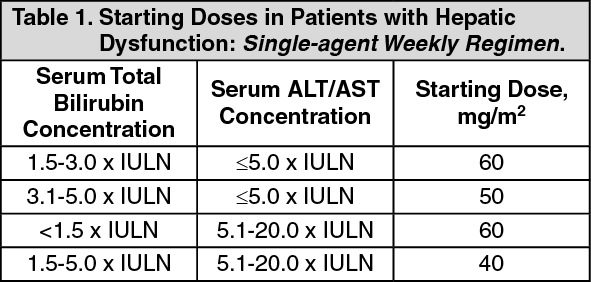
Patients with impaired hepatic function: In patients with hepatic dysfunction, the following starting doses are recommended: (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patients with impaired renal function:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patients with impaired renal function: Studies in this population have not been conducted (See Pharmacology: Pharmacokinetics under Actions). Therefore, caution should be undertaken in patients with impaired renal function. Irinotecan is not recommended for use in patients on dialysis.
Combination-agent dosage schedules: Starting Dose: Irinotecan in Combination with 5-Fluorouracil (5-FU) and folinic acid (FA): Irinotecan in combination with 5-FU and folinic acid is recommended for use in patients with metastatic colorectal cancer. For all regimens, the dose of folinic acid should be administered immediately after irinotecan, with the administration of 5-FU to occur immediately after receipt of folinic acid. The currently recommended regimens are shown as follows.
Regimen 1 (6-week cycle with bolus 5-FU/FA): The recommended starting dose is 125 mg/m
2 of irinotecan, 500 mg/m
2 bolus 5-FU, and 20 mg/m
2 bolus folinic acid.
Regimen 2 (6-week cycle with infusional 5-FU/FA): The recommended starting dose is 180 mg/m
2 of irinotecan, 400 mg/m
2 bolus 5-FU, 600 mg/m
2 5-FU infusion, and 200 mg/m
2 folinic acid.
Lower starting doses may be considered for irinotecan (e.g., 100 mg/m
2) and 5-FU (e.g., 400 mg/m
2) for patients with any of the following conditions: age 65 years and older, prior extensive radiotherapy, performance status of 2, increased bilirubin levels, or gastric cancer. Treatment should be given in repeated 6-week cycles, comprising weekly treatment for 4 weeks, followed by a 2-week rest.
Duration of treatment: For both single-agent and combination-agent regimens, treatment with additional cycles of irinotecan may be continued indefinitely in patients who attain a tumor response or in patients whose cancer remains stable. Patients should be carefully monitored for toxicity and should be removed from therapy if unacceptable toxicity occurs that is not responsive to dose modification and routine supportive care.
Dose modification recommendations: The recommended dose modifications during a cycle of therapy and at the start of each subsequent cycle of therapy for single-agent dosage schedules are described in Table 3. These recommendations are based on toxicities commonly observed with the administration of irinotecan. For modifications at the start of a subsequent cycle of therapy, the dose of irinotecan should be decreased relative to the initial dose of the previous cycle.
The recommended dose modifications during a cycle of therapy and at the start of each subsequent cycle of therapy for irinotecan, 5-FU, and folinic acid are described in Table 4.
All dose modifications should be based on the worst preceding toxicity. A new cycle of therapy should not begin until the toxicity has recovered to Grade 2 or less. Treatment may be delayed 1 to 2 weeks to allow for recovery from treatment-related toxicity. If the patient has not recovered, consideration should be given to discontinuing irinotecan.
Recommended Dose Modifications for Single-agent Schedules: A new cycle of therapy should not begin until the granulocyte count has recovered to ≥1500/mm
3, and the platelet count has recovered to ≥100,000/mm
3, and treatment-related diarrhea is fully resolved. Treatment should be delayed 1 to 2 weeks to allow for recovery from treatment-related toxicities. If the patient has not recovered after a 2-week delay, consideration should be given to discontinuing irinotecan. (See Table 3.)
Click on icon to see table/diagram/image
Recommended Dose Modifications for Irinotecan/5-Fluorouracil/Folinic Acid Combination Schedules: Patients should return to pre-treatment bowel function without requiring antidiarrhea medications for at least 24 hours before the next chemotherapy administration. A new cycle of therapy should not begin until the granulocyte count has recovered to ≥1500/mm
3, and the platelet count has recovered to ≥100,000/mm
3, and treatment-related diarrhea is fully resolved.
Treatment should be delayed 1 to 2 weeks to allow for recovery from treatment-related toxicities. If the patient has not recovered after a 2-week delay, consideration should be given to discontinuing irinotecan. (See Table 4.)
Click on icon to see table/diagram/image


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image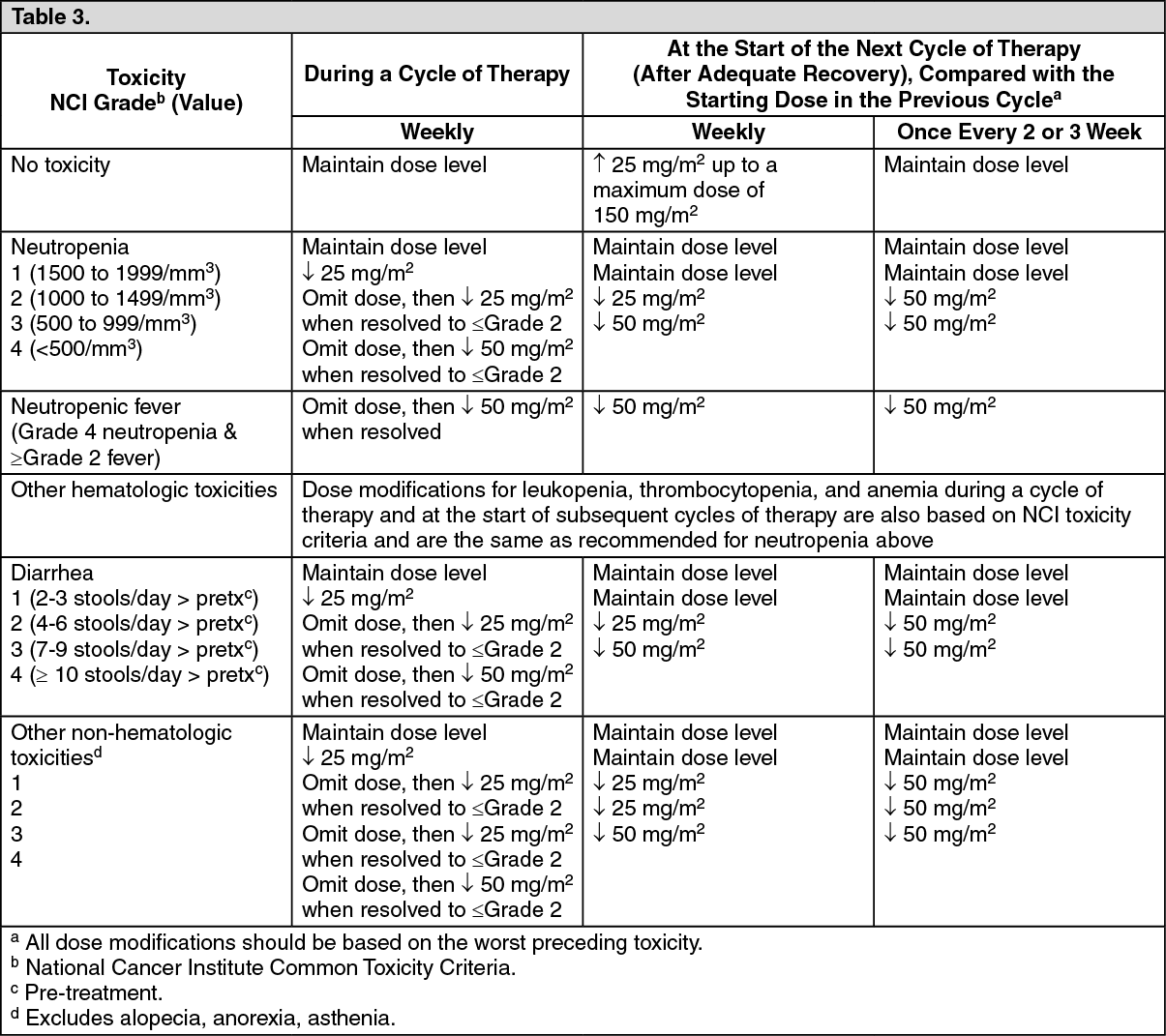 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image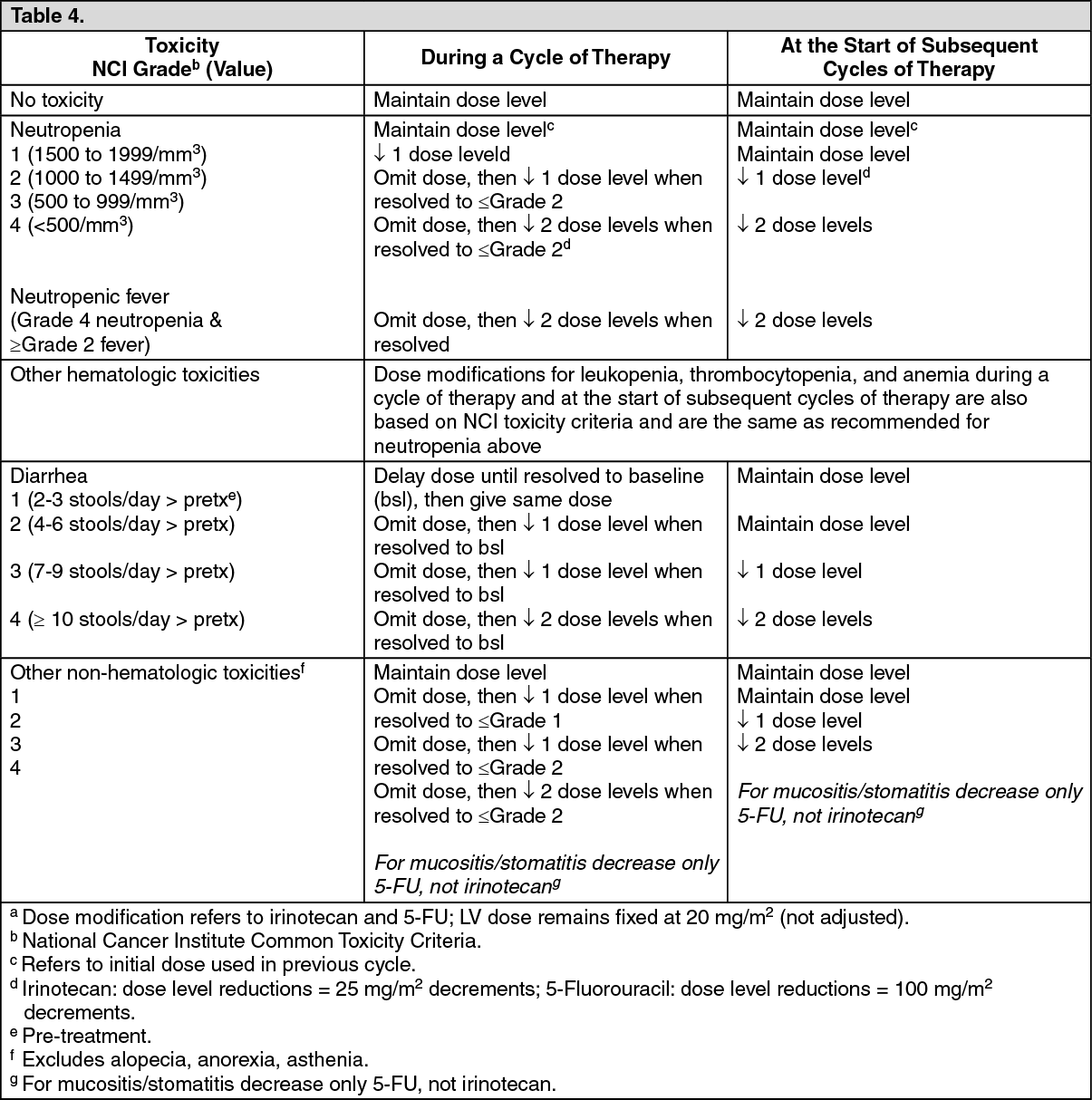 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image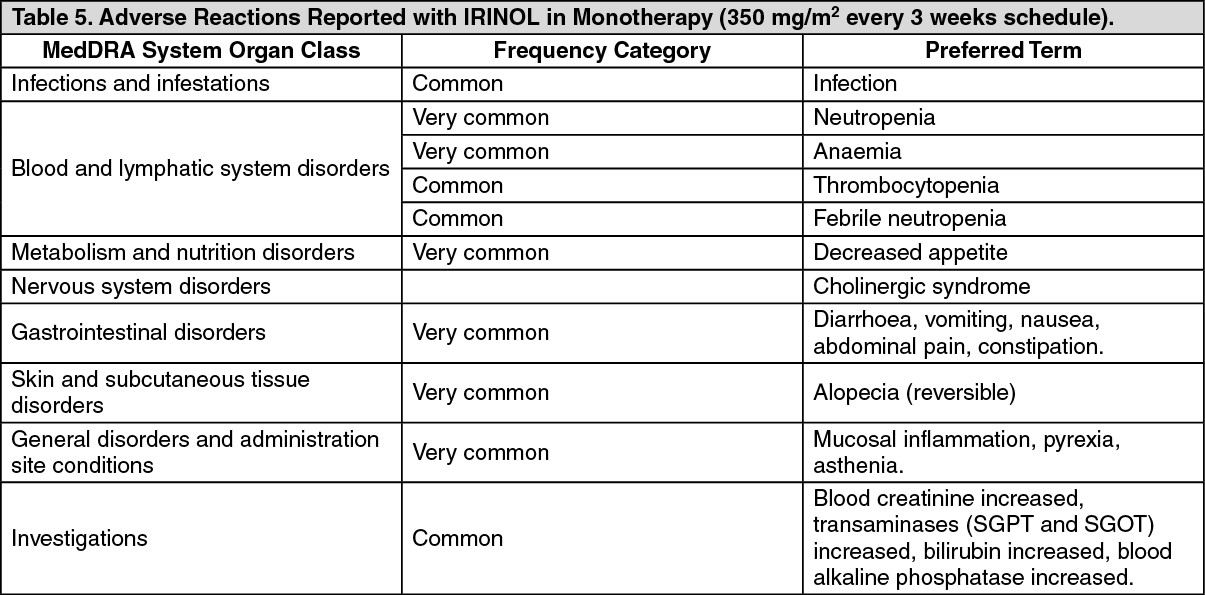 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image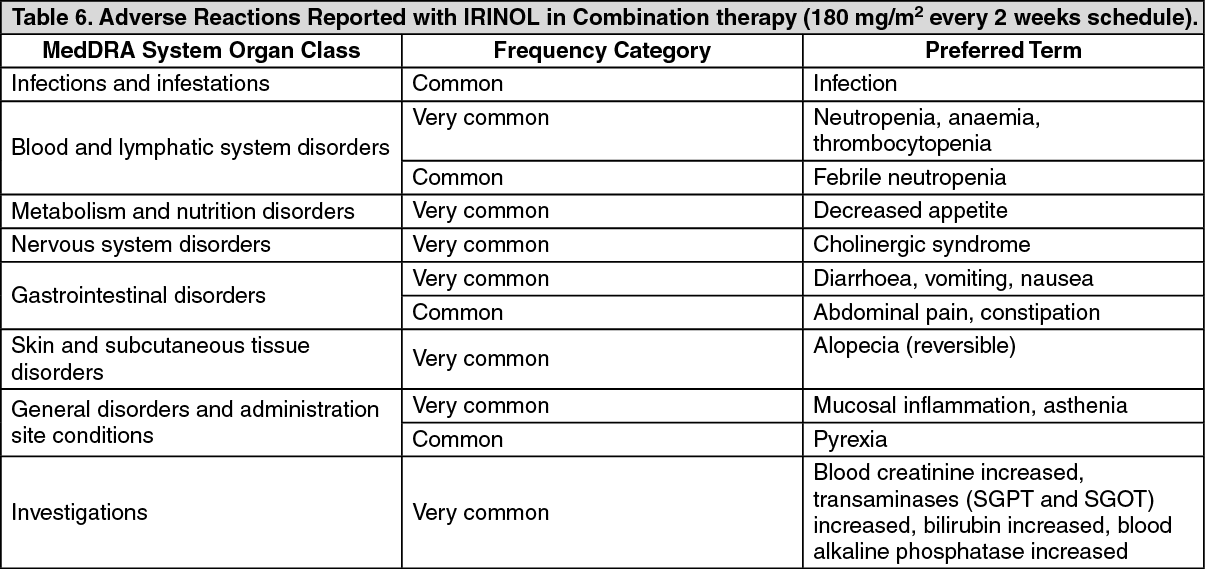 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image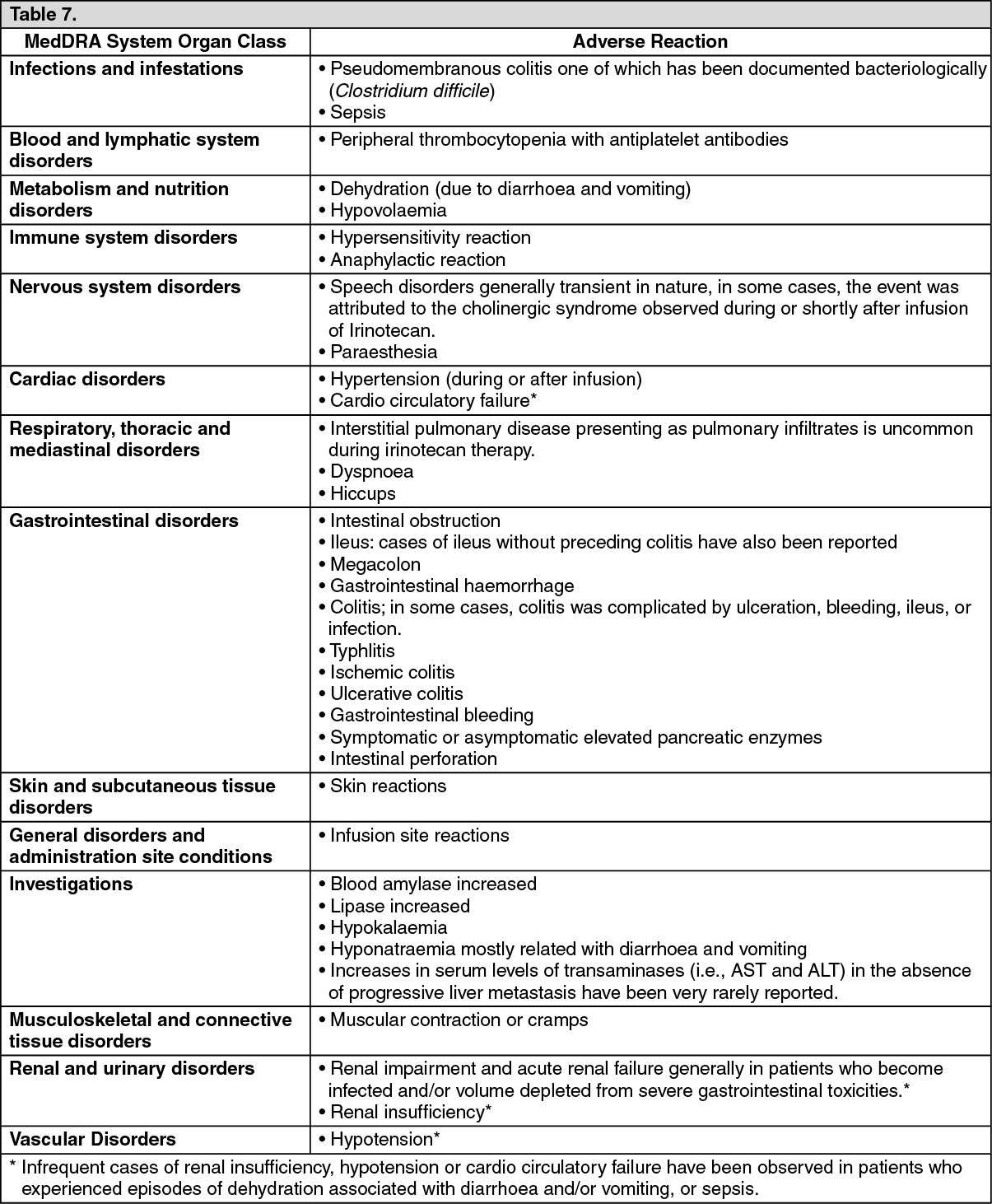 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
