Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors.
ATC code: L01EL01.
Pharmacology: Pharmacodynamics: Mechanism of action: Ibrutinib is a potent, small-molecule inhibitor of Bruton's tyrosine kinase (BTK). Ibrutinib forms a covalent bond with a cysteine residue (Cys-481) in the BTK active site, leading to sustained inhibition of BTK enzymatic activity. BTK, a member of the Tec kinase family, is an important signalling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. The BCR pathway is implicated in the pathogenesis of several B-cell malignancies, including MCL, diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, and CLL. BTK's pivotal role in signalling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis and adhesion. Preclinical studies have shown that ibrutinib effectively inhibits malignant B-cell proliferation and survival in vivo as well as cell migration and substrate adhesion in vitro.
Lymphocytosis: Upon initiation of treatment, a reversible increase in lymphocyte counts (i.e., ≥ 50% increase from baseline and an absolute count > 5,000/mcL), often associated with reduction of lymphadenopathy, has been observed in about three fourths of patients with CLL treated with IMBRUVICA. This effect has also been observed in about one third of patients with relapsed or refractory MCL treated with IMBRUVICA. This observed lymphocytosis is a pharmacodynamic effect and should not be considered progressive disease in the absence of other clinical findings. In both disease types, lymphocytosis typically occurs during the first month of IMBRUVICA therapy and typically resolves within a median of 8.0 weeks in patients with MCL and 14 weeks in patients with CLL. A large increase in the number of circulating lymphocytes (e.g., > 400,000/mcL) has been observed in some patients.
Lymphocytosis was not observed in patients with WM treated with IMBRUVICA.
In vitro platelet aggregation: In an in vitro study, ibrutinib demonstrated inhibition of collagen-induced platelet aggregation. Ibrutinib did not show meaningful inhibition of platelet aggregation using other agonists of platelet aggregation.
Effect on QT/QTc interval and cardiac electrophysiology: The effect of ibrutinib on the QTc interval was evaluated in 20 healthy male and female subjects in a randomised, double-blind thorough QT study with placebo and positive controls. At a supratherapeutic dose of 1,680 mg, ibrutinib did not prolong the QTc interval to any clinically relevant extent. The largest upper bound of the 2-sided 90% CI for the baseline adjusted mean differences between ibrutinib and placebo was below 10 ms. In this same study, a concentration dependent shortening in the QTc interval was observed (-5.3 ms [90% CI: -9.4, -1.1] at a C
max of 719 ng/mL following the supratherapeutic dose of 1,680 mg).
Clinical efficacy and safety: MCL: The safety and efficacy of IMBRUVICA in patients with relapsed or refractory MCL were evaluated in a single open-label, multi-center phase 2 study (PCYC-1104-CA) of 111 patients. The median age was 68 years (range: 40 to 84 years), 77% were male and 92% were Caucasian. Patients with Eastern Cooperative Oncology Group (ECOG) performance status of 3 or greater were excluded from the study. The median time since diagnosis was 42 months, and median number of prior treatments was 3 (range: 1 to 5 treatments), including 35% with prior high-dose chemotherapy, 43% with prior bortezomib, 24% with prior lenalidomide, and 11% with prior autologous or allogeneic stem cell transplant. At baseline, 39% of patients had bulky disease (≥ 5 cm), 49% had high-risk score by Simplified MCL International Prognostic Index (MIPI), and 72% had advanced disease (extranodal and/or bone marrow involvement) at screening.
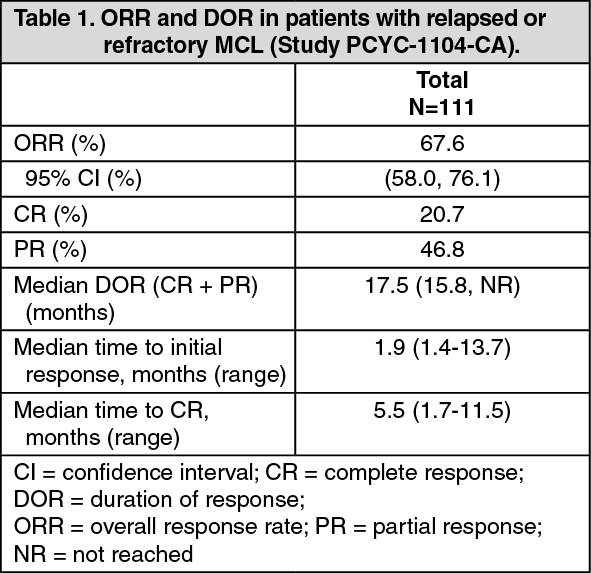
IMBRUVICA was administered orally at 560 mg once daily until disease progression or unacceptable toxicity. Tumour response was assessed according to the revised International Working Group (IWG) for non-Hodgkin's lymphoma (NHL) criteria. The primary endpoint in this study was investigator-assessed overall response rate (ORR). Responses to IMBRUVICA are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The efficacy data was further evaluated by an Independent Review Committee (IRC) demonstrating an ORR of 69%, with a 21% complete response (CR) rate and a 48% partial response (PR) rate. The IRC estimated median DOR was 19.6 months.
The overall response to IMBRUVICA was independent of prior treatment including bortezomib and lenalidomide or underlying risk/prognostic factors, bulky disease, gender or age.
The safety and efficacy of IMBRUVICA were demonstrated in a randomised phase 3, open-label, multicenter study including 280 patients with MCL who received at least one prior therapy (Study MCL3001). Patients were randomised 1:1 to receive either IMBRUVICA orally at 560 mg once daily for 21 days or temsirolimus intravenously at 175 mg on Days 1, 8, 15 of the first cycle followed by 75 mg on Days 1, 8, 15 of each subsequent 21-day cycle. Treatment on both arms continued until disease progression or unacceptable toxicity. The median age was 68 years (range, 34; 88 years), 74% were male and 87% were Caucasian. The median time since diagnosis was 43 months, and median number of prior treatments was 2 (range: 1 to 9 treatments), including 51% with prior high-dose chemotherapy, 18% with prior bortezomib, 5% with prior lenalidomide, and 24% with prior stem cell transplant. At baseline, 53% of patients had bulky disease (≥ 5 cm), 21% had high-risk score by Simplified MIPI, 60% had extranodal disease and 54% had bone marrow involvement at screening.
Progression-free survival (PFS) was assessed by IRC according to the revised International Working Group (IWG) for non-Hodgkin's lymphoma (NHL) criteria. Efficacy results for Study MCL3001 are shown in Table 2 and the Kaplan-Meier curve for PFS in Figure 1. (See Table 2.)
Click on icon to see table/diagram/image
A smaller proportion of patients treated with ibrutinib experienced a clinically meaningful worsening of lymphoma symptoms versus temsirolimus (27% versus 52%) and time to worsening of symptoms occurred more slowly with ibrutinib versus temsirolimus (HR 0.27, p < 0.0001). (See Figure 1.)
Click on icon to see table/diagram/image
CLL: Patients previously untreated for CLL: Single agent: A randomised, multicenter, open-label phase 3 study (PCYC-1115-CA) of IMBRUVICA versus chlorambucil was conducted in patients with treatment-naïve CLL who were 65 years of age or older. Patients between 65 and 70 years of age were required to have at least one comorbidity that precluded the use of frontline chemo-immunotherapy with fludarabine, cyclophosphamide, and rituximab. Patients (n = 269) were randomised 1:1 to receive either IMBRUVICA 420 mg daily until disease progression or unacceptable toxicity, or chlorambucil at a starting dose of 0.5 mg/kg on days 1 and 15 of each 28-day cycle for a maximum of 12 cycles, with an allowance for intrapatient dose increases up to 0.8 mg/kg based on tolerability. After confirmed disease progression, patients on chlorambucil were able to crossover to ibrutinib.
The median age was 73 years (range, 65 to 90 years), 63% were male, and 91% were Caucasian. Ninety one percent of patients had a baseline ECOG performance status of 0 or 1 and 9% had an ECOG performance status of 2. The study enrolled 269 patients with CLL. At baseline, 45% had advanced clinical stage (Rai Stage III or IV), 35% of patients had at least one tumor ≥ 5 cm, 39% with baseline anaemia, 23% with baseline thrombocytopenia, 65% had elevated β2 microglobulin > 3500 mcg/L, 47% had a CrCL < 60 mL/min, 20% of patients presented with del11q, 6% of patients presented with del 17p/tumor protein 53 (TP53) mutation, and 44% of patients presented with unmutated immunoglobulin heavy chain variable region (IGHV).
Progression free survival (PFS) as assessed by IRC according to International Workshop on CLL (IWCLL) criteria indicated an 84% statistically significant reduction in the risk of death or progression in the IMBRUVICA arm. Efficacy results for Study PCYC-1115-CA are shown in Table 3 and the Kaplan-Meier curves for PFS and OS are shown in Figures 2 and 3, respectively.
There was a statistically significant sustained platelet or haemoglobin improvement in the ITT population in favor of ibrutinib versus chlorambucil. In patients with baseline cytopenias, sustained haematologic improvement was: platelets 77.1% versus 42.9%; haemoglobin 84.3% versus 45.5% for ibrutinib and chlorambucil respectively. (See Table 3 and Figures 2 and 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
48-month follow up: With a median follow-up time on study of 48 months in Study PCYC-1115-CA and its extension study, an 86% reduction in the risk of death or progression by investigator assessment was observed for patients in the IMBRUVICA arm. The median investigator-assessed PFS was not reached in the IMBRUVICA arm and was 15 months [95% CI (10.22, 19.35)] in the chlorambucil arm; (HR=0.14 [95% CI (0.09, 0.21)]). The 4-year PFS estimate was 73.9% in the IMBRUVICA arm and 15.5% in the chlorambucil arm, respectively. The updated Kaplan-Meier curve for PFS is shown in Figure 4.
The investigator-assessed ORR was 91.2% in the IMBRUVICA arm versus 36.8% in the chlorambucil arm. The CR rate according to IWCLL criteria was 16.2% in the IMBRUVICA arm versus 3.0% in the chlorambucil arm. At the time of long-term follow-up, a total of 73 subjects (54.9%) originally randomised to the chlorambucil arm subsequently received ibrutinib as cross-over treatment. The Kaplan-Meier landmark estimate for OS at 48-months was 85.5% in the IMBRUVICA arm.
The treatment effect of ibrutinib in Study PCYC-1115-CA was consistent across high-risk patients with del 17p/TP53 mutation, del 11q, and/or unmutated IGHV. (See Figure 4.)
Click on icon to see table/diagram/image
Combination therapy: The safety and efficacy of IMBRUVICA in patients with previously untreated CLL/SLL were further evaluated in a randomized, multi-center, open-label, Phase 3 study (PCYC-1130-CA) of IMBRUVICA in combination with obinutuzumab versus chlorambucil in combination with obinutuzumab. The study enrolled patients who were 65 years of age or older or <65 years of age with coexisting medical conditions, reduced renal function as measured by creatinine clearance <70 mL/min, or presence of del17p/TP53 mutation. Patients (n=229) were randomized 1:1 to receive either IMBRUVICA 420 mg daily until disease progression or unacceptable toxicity or chlorambucil at a dose of 0.5 mg/kg on Days 1 and 15 of each 28-day cycle for 6 cycles. In both arms, patients received 1000 mg of obinutuzumab on Days 1, 8 and 15 of the first cycle, followed by treatment on the first day of 5 subsequent cycles (total of 6 cycles, 28 days each). The first dose of obinutuzumab was divided between day 1 (100 mg) and day 2 (900 mg).
The median age was 71 years (range, 40 to 87 years), 64% were male, and 96% were Caucasian. All patients had a baseline ECOG performance status of 0 (48%) or 1-2 (52%). At baseline, 52% had advanced clinical stage (Rai Stage III or IV), 32% of patients had bulky disease (≥5 cm), 44% with baseline anaemia, 22% with baseline thrombocytopenia, 28% had a CrCL <60 mL/min, and the median Cumulative Illness Rating Score for Geriatrics (CIRS-G) was 4 (range, 0 to 12). At baseline, 65% of patients presented with CLL/SLL with high risk factors (del17p/TP53 mutation [18%], del11q [15%], or unmutated IGHV [54%]).
Progression free survival (PFS) was assessed by IRC according to IWCLL criteria indicated a 77% statistically significant reduction in the risk of death or progression in the IMBRUVICA arm. With a median follow up time on study of 31 months, the median PFS was not reached in the IMBRUVICA+obinutuzumab arm and was 19 months in the chlorambucil+obinutuzumab arm. Efficacy results for Study PCYC 1130 CA are shown in Table 4 and the Kaplan-Meier curve for PFS is shown in Figure 5. (See Table 4 and Figure 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The treatment effect of ibrutinib was consistent across the high-risk CLL/SLL population (del17p/TP53 mutation, del11q, or unmutated IGHV), with a PFS HR of 0.15 [95% CI (0.09, 0.27)], as shown in Table 5. The 2-year PFS rate estimates for the high-risk CLL/SLL population were 78.8% [95% CI (67.3, 86.7)] and 15.5% [95% CI (8.1, 25.2)] in the IMBRUVICA+obinutuzumab and chlorambucil+obinutuzumab arms, respectively. (See Table 5.)
Click on icon to see table/diagram/image
Any grade infusion-related reactions were observed in 25% of patients treated with IMBRUVICA+obinutuzumab and 58% of patients treated with chlorambucil+obinutuzumab. Grade 3 or higher or serious infusion-related reactions were observed in 3% of patients treated with IMBRUVICA+obinutuzumab and 9% of patients treated with chlorambucil+obinutuzumab.
The safety and efficacy of IMBRUVICA in patients with previously untreated CLL or SLL were further evaluated in a randomised, multi-center, open-label, phase 3 study (E1912) of IMBRUVICA in combination with rituximab (IR) versus standard fludarabine, cyclophosphamide, and rituximab (FCR) chemo-immunotherapy. The study enrolled previously untreated patients with CLL or SLL who were 70 years or younger. Patients with del17p were excluded from the study. Patients (n=529) were randomised 2:1 to receive either IR or FCR. IMBRUVICA was administered at a dose of 420 mg daily until disease progression or unacceptable toxicity. Fludarabine was administered at a dose of 25 mg/m2, and cyclophosphamide was administered at a dose of 250 mg/m2, both on Days 1, 2, and 3 of Cycles 1-6. Rituximab was initiated in Cycle 2 for the IR arm and in Cycle 1 for the FCR arm and was administered at a dose of 50 mg/m2 on Day 1 of the first cycle, 325 mg/m2 on Day 2 of the first cycle, and 500 mg/m2 on Day 1 of 5 subsequent cycles, for a total of 6 cycles. Each cycle was 28 days.
The median age was 58 years (range, 28 to 70 years), 67% were male, and 90% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1 (98%) or 2 (2%). At baseline, 43% of patients presented with Rai Stage III or IV, and 59% of patients presented with CLL/SLL with high risk factors (TP53 mutation [6%], del11q [22%], or unmutated IGHV [53%]).
With a median follow-up time on study of 37 months, efficacy results for E1912 are shown in Table 6. The Kaplan-Meier curves for PFS, assessed according to IWCLL criteria, and OS are shown in Figures 6 and 7, respectively. (See Table 6 and Figure 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The treatment effect of ibrutinib was consistent across the high-risk CLL/SLL population (TP53 mutation, del11q, or unmutated IGHV), with a PFS HR of 0.23 [95% CI (0.13, 0.40)], p <0.0001, as shown in Table 7. The 3-year PFS rate estimates for the high-risk CLL/SLL population were 90.4% [95% CI (85.4, 93.7)] and 60.3% [95% CI (46.2, 71.8)] in the IR and FCR arms, respectively. (See Table 7 and Figure 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patients with CLL who received at least one prior therapy: Single agent: The safety and efficacy of IMBRUVICA in patients with CLL were demonstrated in one uncontrolled study and one randomised, controlled study. The open-label, multi-center study (PCYC-1102-CA) included 51 patients with relapsed or refractory CLL, who received 420 mg once daily. IMBRUVICA was administered until disease progression or unacceptable toxicity. The median age was 68 years (range: 37 to 82 years), median time since diagnosis was 80 months, and median number of prior treatments was 4 (range: 1 to 12 treatments), including 92.2% with a prior nucleoside analog, 98.0% with prior rituximab, 86.3% with a prior alkylator, 39.2% with prior bendamustine and 19.6% with prior ofatumumab. At baseline, 39.2% of patients had Rai Stage IV, 45.1% had bulky disease (≥ 5 cm), 35.3% had deletion 17p and 31.4% had deletion 11q.
ORR was assessed according to the 2008 IWCLL criteria by investigators and IRC. At a median duration follow up of 16.4 months, the ORR by IRC for the 51 relapsed or refractory patients was 64.7% (95% CI: 50.1%; 77.6%), all PRs. The ORR including PR with lymphocytosis was 70.6%. Median time to response was 1.9 months. The DOR ranged from 3.9 to 24.2+ months. The median DOR was not reached.
A randomised, multi-center, open-label phase 3 study of IMBRUVICA versus ofatumumab (PCYC-1112-CA) was conducted in patients with relapsed or refractory CLL. Patients (n = 391) were randomised 1:1 to receive either IMBRUVICA 420 mg daily until disease progression or unacceptable toxicity, or ofatumumab for up to 12 doses (300/2,000 mg). Fifty-seven patients randomised to ofatumumab crossed over following progression to receive IMBRUVICA. The median age was 67 years (range: 30 to 88 years), 68% were male, and 90% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 91 months and the median number of prior treatments was 2 (range: 1 to 13 treatments). At baseline, 58% of patients had at least one tumour ≥5 cm. Thirty-two percent of patients had deletion 17p (with 50% of patients having deletion 17p/TP53 mutation), 24% had 11q deletion, and 47% of patients had unmutated IGHV.
Progression free survival (PFS) as assessed by an IRC according to IWCLL criteria indicated a 78% statistically significant reduction in the risk of death or progression for patients in the IMBRUVICA arm. Analysis of OS demonstrated a 57% statistically significant reduction in the risk of death for patients in the IMBRUVICA arm. Efficacy results for Study PCYC-1112-CA are shown in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
The efficacy was similar across all of the subgroups examined, including in patients with and without deletion 17p, a pre-specified stratification factor (Table 9). (See Table 9.)
Click on icon to see table/diagram/image
The Kaplan-Meier curve for PFS is shown in Figure 8. (See Figure 8.)
Click on icon to see table/diagram/image
Final Analysis at 65-month follow-up: With a median follow-up time on study of 65 months in Study PCYC-1112-CA, an 85% reduction in the risk of death or progression by investigator assessment was observed for patients in the IMBRUVICA arm. The median investigator-assessed PFS according to IWCLL criteria was 44.1 months [95% CI (38.47, 56.18)] in the IMBRUVICA arm and 8.1 months [95% CI (7.79, 8.25)] in the ofatumumab arm, respectively; HR=0.15 [95% CI (0.11, 0.20)]. The updated Kaplan-Meier curve for PFS is shown in Figure 9. The investigator-assessed ORR in the IMBRUVICA arm was 87.7% versus 22.4% in the ofatumumab arm. At the time of final analysis, 133 (67.9%) of the 196 subjects originally randomized to the ofatumumab treatment arm had crossed over to ibrutinib treatment. The median investigator-assessed PFS2 (time from randomisation until PFS event after first subsequent anti-neoplastic therapy) according to IWCLL criteria was 65.4 months [95% CI (51.61, not estimable)] in the IMBRUVICA arm and 38.5 months [95% CI (19.98, 47.24)] in the ofatumumab arm, respectively; HR=0.54 [95% CI (0.41, 0.71)]. The median OS was 67.7 months [95% CI (61.0, not estimable)] in the IMBRUVICA arm.
The treatment effect of ibrutinib in Study PCYC-1112-CA was consistent across high-risk patients with deletion 17p/TP53 mutation, deletion 11q, and/or unmutated IGHV. (See Figure 9.)
Click on icon to see table/diagram/image
Combination therapy: The safety and efficacy of IMBRUVICA in patients previously treated for CLL were further evaluated in a randomised, multicenter, double-blinded phase 3 study of IMBRUVICA in combination with BR versus placebo+BR (Study CLL3001). Patients (n=578) were randomised 1:1 to receive either IMBRUVICA 420 mg daily or placebo in combination with BR until disease progression, or unacceptable toxicity. All patients received BR for a maximum of six 28-day cycles. Bendamustine was dosed at 70 mg/m
2 infused IV over 30 minutes on Cycle 1, Days 2 and 3, and on Cycles 2-6, Days 1 and 2 for up to 6 cycles. Rituximab was administered at a dose of 375 mg/m
2 in the first cycle, Day 1, and 500 mg/m
2 Cycles 2 through 6, Day 1. Ninety patients randomised to placebo+BR crossed over to receive IMBRUVICA following IRC confirmed progression. The median age was 64 years (range, 31 to 86 years), 66% were male, and 91% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 6 years and the median number of prior treatments was 2 (range, 1 to 11 treatments). At baseline, 56% of patients had at least one tumour ≥5 cm, 26% had del11q.
Progression free survival (PFS) was assessed by IRC according to IWCLL criteria. Efficacy results for Study CLL3001 are shown in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
WM: Single agent: The safety and efficacy of IMBRUVICA in WM (IgM-excreting lymphoplasmacytic lymphoma) were evaluated in an open-label, multi-center, single-arm trial of 63 previously treated patients. The median age was 63 years (range: 44 to 86 years), 76% were male, and 95% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 74 months, and the median number of prior treatments was 2 (range: 1 to 11 treatments). At baseline, the median serum IgM value was 3.5 g/dL, and 60% of patients were anaemic (haemoglobin ≤11 g/dL or 6.8 mmol/L).
IMBRUVICA was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The primary endpoint in this study was ORR per investigator assessment. The ORR and DOR were assessed using criteria adopted from the Third International Workshop of WM. Responses to IMBRUVICA are shown in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
The median time to response was 1.0 month (range: 0.7-13.4 months).
Efficacy results were also assessed by an IRC demonstrating an ORR of 83%, with a 11% VGPR rate and a 51% PR rate.
Combination therapy: The safety and efficacy of IMBRUVICA in WM were further evaluated in patients with treatment-naïve or previously treated WM in a randomised, multicenter, double-blinded phase 3 study of IMBRUVICA in combination with rituximab versus placebo in combination with rituximab (PCYC-1127-CA). Patients (n=150) were randomised 1:1 to receive either IMBRUVICA 420 mg daily or placebo in combination with rituximab until disease progression or unacceptable toxicity. Rituximab was administered weekly at a dose of 375 mg/m2 for 4 consecutive weeks (weeks 1-4) followed by a second course of weekly rituximab for 4 consecutive weeks (weeks 17-20).
The median age was 69 years (range, 36 to 89 years), 66% were male, and 79% were Caucasian. Ninety-three percent of patients had a baseline ECOG performance status of 0 or 1, and 7% of patients had a baseline ECOG performance status of 2. Forty-five percent of patients were treatment-naïve, and 55% of patients were previously treated. The median time since diagnosis was 52.6 months (treatment-naïve patients=6.5 months and previously treated patients=94.3 months). Among previously treated patients, the median number of prior treatments was 2 (range, 1 to 6 treatments). At baseline, the median serum IgM value was 3.2 g/dL (range, 0.6 to 8.3 g/dL), 63% of patients were anaemic (haemoglobin ≤11 g/dL or 6.8 mmol/L) and MYD88 L265P mutations were present in 77% of patients, absent in 13% of patients, and 9% of patients were not evaluable for mutation status.
At the primary analysis, with a median follow-up of 26.5 months, the IRC-assessed PFS hazard ratio was 0.20 [95% CI (0.11, 0.38)]. PFS hazard ratios for treatment-naïve patients, previously treated patients, and patients with or without MYD88 L265P mutations were consistent with the PFS hazard ratio for the ITT population.
Grade 3 or 4 infusion-related reactions were observed in 1% of patients treated with IMBRUVICA+rituximab and 16% of patients treated with placebo+rituximab.
Tumor flare in the form of IgM increase occurred in 8.0% of subjects in the IMBRUVICA+rituximab arm and 46.7% of subjects in the placebo+rituximab arm.
Final Analysis at 63-month follow-up: With an overall follow-up of 63 months, efficacy results as assessed by an IRC at the time of the final analysis for PCYC-1127-CA are shown in Table 12 and the Kaplan-Meier curve for PFS is shown in Figure 10. PFS hazard ratios for treatment-naïve patients (0.31 [95% CI (0.14, 0.69)]) and previously treated patients (0.22 [95% CI (0.11, 0.43)]) were consistent with the PFS hazard ratio for the ITT population. (See Table 12 and Figure 10.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Study PCYC-1127-CA had a separate monotherapy arm of 31 patients with previously treated WM who failed prior rituximab-containing therapy and received single agent IMBRUVICA. The median age was 67 years (range, 47 to 90 years). Eighty-one percent of patients had a baseline ECOG performance status of 0 or 1, and 19% had a baseline ECOG performance status of 2. The median number of prior treatments was 4 (range, 1 to 7 treatments). With an overall follow-up of 61 months, the response rate observed in Study PCYC-1127-CA monotherapy arm per IRC assessment was 77% (0% CR, 29% VGPR, 48% PR). The median duration of response was 33 months (range, 2.4 to 60.2+ months). The overall response rate per IRC observed in the monotherapy arm was 87% (0% CR, 29% VGPR, 48% PR, 10% MR). The median duration of overall response was 39 months (range, 2.07 to 60.2+ months).
Chronic graft versus host disease (cGVHD): The safety and efficacy of IMBRUVICA in cGVHD were evaluated in an open-label, multi-center, single-arm trial of 42 patients with cGVHD after failure of first line corticosteroid therapy and required additional therapy. The median age was 56 years (range, 19 to 74 years), 52% were male, and 93% were Caucasian. The most common underlying malignancies leading to transplant were acute lymphocytic leukemia, acute myeloid leukemia, and CLL. The median time since diagnosis was 14 months and the median number of prior cGVHD treatments was 2 (range, 1 to 3 treatments). The majority of patients (88%) had at least 2 organs involved at baseline, with the most commonly involved organs being mouth (86%), skin (81%), and gastrointestinal tract (33%). The median daily steroid dose per body weight at baseline was 0.3 mg/kg/day.
IMBRUVICA was administered orally at 420 mg once daily until disease progression, unacceptable toxicity or recurrence of underlying malignancy. The primary endpoint in this study was best ORR per investigator assessment using the 2005 National Institutes of Health Consensus Panel Response Criteria with modification. Responses were seen across involved organs for cGVHD (skin, mouth, gastrointestinal tract, and liver). Efficacy results are shown in Table 13. (See Table 13.)
Click on icon to see table/diagram/image
ORR results were supported by exploratory analyses of patient-reported symptom burden which showed at least a 7-point decrease in Lee Symptom Scale overall summary score in 24% (10/42) of patients on at least 2 consecutive visits.
Pharmacokinetics: Absorption: Ibrutinib is rapidly absorbed after oral administration with a median T
max of 1 to 2 hours. Absolute bioavailability in fasted condition (n = 8) was 2.9% (90% CI = 2.1 - 3.9) and doubled when combined with a meal. Pharmacokinetics of ibrutinib does not significantly differ in patients with different B-cell malignancies. Ibrutinib exposure increases with doses up to 840 mg. The steady state AUC observed in patients at 560 mg is (mean ± standard deviation) 953 ± 705 ng h/mL and in patients at 420mg with cGVHD is 1159 ± 583 ng·h/mL. Administration of ibrutinib in fasted condition resulted in approximately 60% of exposure (AUC
last) as compared to either 30 minutes before, 30 minutes after (fed condition) or 2 hours after a high fat breakfast.
Ibrutinib has a pH dependent solubility, with lower solubility at higher pH. In fasted healthy subjects administered a single 560 mg dose of ibrutinib after taking omeprazole at 40 mg once daily for 5 days, compared to ibrutinib alone, geometric mean ratios (90% CI) were 83% (68-102%), 92% (78-110%), and 38% (26-53%) for AUC
0-24, AUC
last, and C
max, respectively.
Distribution: Reversible binding of ibrutinib to human plasma protein in vitro was 97.3% with no concentration dependence in the range of 50 to 1,000 ng/mL. The apparent volume of distribution at steady state (V
d,
ss/F) was approximately 10,000 L.
Metabolism: Ibrutinib is metabolised primarily by CYP3A4 to produce a dihydrodiol metabolite with an inhibitory activity towards BTK approximately 15 times lower than that of ibrutinib. Involvement of CYP2D6 in the metabolism of ibrutinib appears to be minimal.
Therefore, no precautions are necessary in patients with different CYP2D6 genotypes.
Elimination: Apparent clearance (CL/F) is approximately 1,000 L/h. The half-life of ibrutinib is 4 to 13 hours. After a single oral administration of radiolabeled [
14C]-ibrutinib in healthy subjects, approximately 90% of radioactivity was excreted within 168 hours, with the majority (80%) excreted in the faeces and < 10% accounted for in urine. Unchanged ibrutinib accounted for approximately 1% of the radiolabeled excretion product in faeces and none in urine.
Special populations: Elderly: Population pharmacokinetics indicated that age does not significantly influence ibrutinib clearance from the circulation.
Paediatric population: No pharmacokinetic studies were performed with IMBRUVICA in patients under 18 years of age.
Gender: Population pharmacokinetics data indicated that gender does not significantly influence ibrutinib clearance from the circulation.
Race: There are insufficient data to evaluate the potential effect of race on ibrutinib pharmacokinetics.
Body weight: Population pharmacokinetics data indicated that body weight (range: 41-146 kg; mean [SD]: 83 [19 kg]) had a negligible effect on ibrutinib clearance.
Renal impairment: Ibrutinib has minimal renal clearance; urinary excretion of metabolites is < 10% of the dose. No specific studies have been conducted to date in subjects with impaired renal function. There are no data in patients with severe renal impairment or patients on dialysis (see Dosage & Administration).
Hepatic impairment: Ibrutinib is metabolised in the liver. A hepatic impairment trial was performed in non-cancer subjects administered a single dose of 140 mg of medicinal product under fasting conditions. The effect of impaired liver function varied substantially between individuals, but on average a 2.7-, 8.2-, and 9.8-fold increase in ibrutinib exposure (AUC
last) was observed in subjects with mild (n = 6, Child-Pugh class A), moderate (n = 10, Child-Pugh class B) and severe (n = 8, Child-Pugh class C) hepatic impairment, respectively. The free fraction of ibrutinib also increased with degree of impairment, with 3.0, 3.8 and 4.8% in subjects with mild, moderate and severe liver impairment, respectively, compared to 3.3% in plasma from matched healthy controls within this study. The corresponding increase in unbound ibrutinib exposure (AUC
unbound, last) is estimated to be 4.1-, 9.8-, and 13-fold in subjects with mild, moderate, and severe hepatic impairment, respectively (see Dosage & Administration).
Co-administration with transport substrates/inhibitors:
In vitro studies indicated that ibrutinib is not a substrate of P-gp, nor other major transporters, except OCT2. The dihydrodiol metabolite and other metabolites are P-gp substrates. Ibrutinib is an
in vitro inhibitor of P-gp and BCRP (see Interactions).
Toxicology: Preclinical safety data: The following adverse effects were seen in studies of 13-weeks duration in rats and dogs. Ibrutinib was found to induce gastrointestinal effects (soft faeces/diarrhoea and/or inflammation) and lymphoid depletion in rats and dogs with a No Observed Adverse Effect Level (NOAEL) of 30 mg/kg/day in both species. Based on mean exposure (AUC) at the 560 mg/day clinical dose, AUC ratios were 2.6 and 21 at the NOAEL in male and female rats, and 0.4 and 1.8 at the NOAEL in male and female dogs, respectively. Lowest Observed Effect Level (LOEL) (60 mg/kg/day) margins in the dog are 3.6-fold (males) and 2.3-fold (females). In rats, moderate pancreatic acinar cell atrophy (considered adverse) was observed at doses of ≥ 100 mg/kg in male rats (AUC exposure margin of 2.6-fold) and not observed in females at doses up to 300 mg/kg/day (AUC exposure margin of 21.3-fold). Mildly decreased trabecular and cortical bone was seen in female rats administered ≥ 100 mg/kg/day (AUC exposure margin of 20.3-fold). All gastrointestinal, lymphoid and bone findings recovered following recovery periods of 6-13 weeks. Pancreatic findings partially recovered during comparable reversal periods.
Juvenile toxicity studies have not been conducted.
Carcinogenicity/genotoxicity: Ibrutinib was not carcinogenic in a 6-month study in the transgenic (Tg.rasH2) mouse at oral doses up to 2000 mg/kg/day with an exposure margin of approximately 23 (males) to 37 (females) times the human AUC of ibrutinib at a dose of 560 mg daily.
Ibrutinib has no genotoxic properties when tested in bacteria, mammalian cells or in mice.
Reproductive toxicity: In pregnant rats, ibrutinib at a dose of 80 mg/kg/day was associated with increased post-implantation loss and increased visceral (heart and major vessels) malformations and skeletal variations with an exposure margin 14 times the AUC found in patients at a daily dose of 560 mg. At a dose of ≥ 40 mg/kg/day, ibrutinib was associated with decreased foetal weights (AUC ratio of ≥ 5.6 as compared to daily dose of 560 mg in patients). Consequently the foetal NOAEL was 10 mg/kg/day (approximately 1.3 times the AUC of ibrutinib at a dose of 560 mg daily) (see Use in Pregnancy & Lactation).
In pregnant rabbits, ibrutinib at a dose of 15 mg/kg/day or greater was associated with skeletal malformations (fused sternebrae) and ibrutinib at a dose of 45 mg/kg/day was associated with increased post-implantation loss. Ibrutinib caused malformations in rabbits at a dose of 15 mg/kg/day (approximately 2.0 times the exposure (AUC) in patients with MCL administered ibrutinib 560 mg daily and 2.8 times the exposure in patients with CLL or WM receiving ibrutinib dose 420 mg per day). Consequently the foetal NOAEL was 5 mg/kg/day (approximately 0.7 times the AUC of ibrutinib at a dose of 560 mg daily) (see Use in Pregnancy & Lactation).
Fertility: No effects on fertility or reproductive capacities were observed in male or female rats up to the maximum dose tested, 100 mg/kg/day (HED 16 mg/kg/day).



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image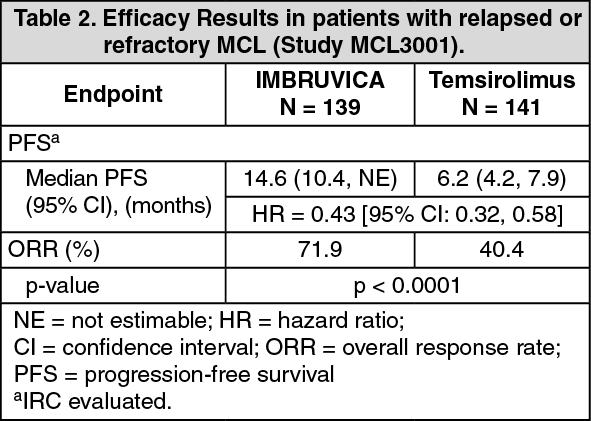 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image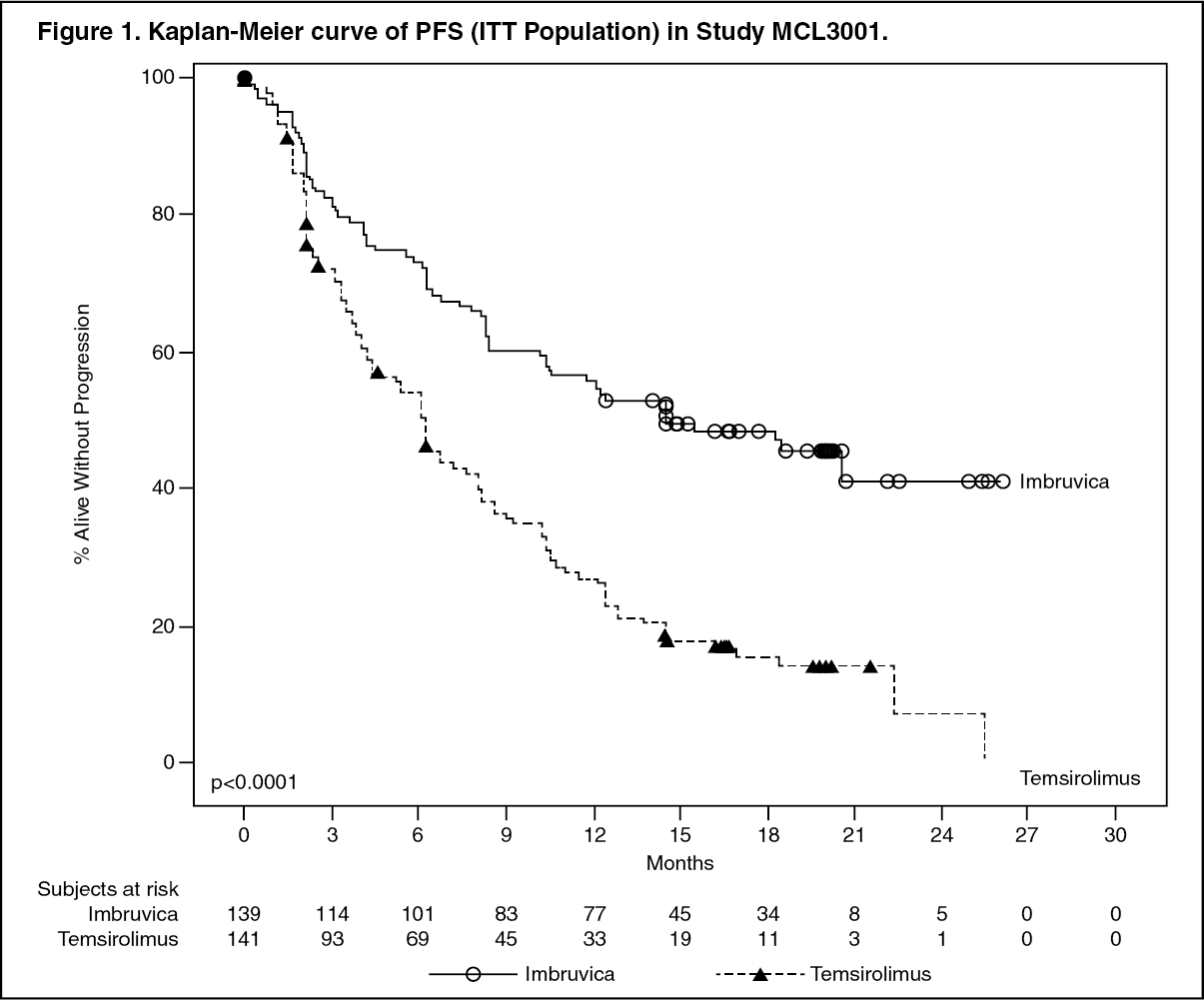 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image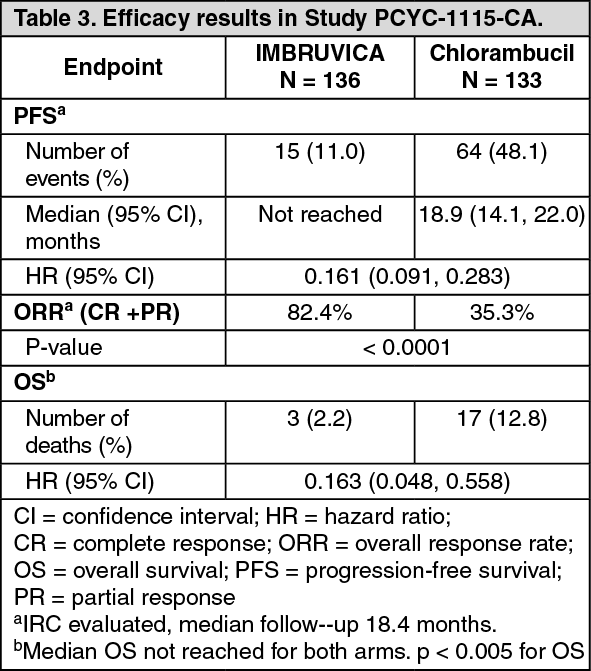 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image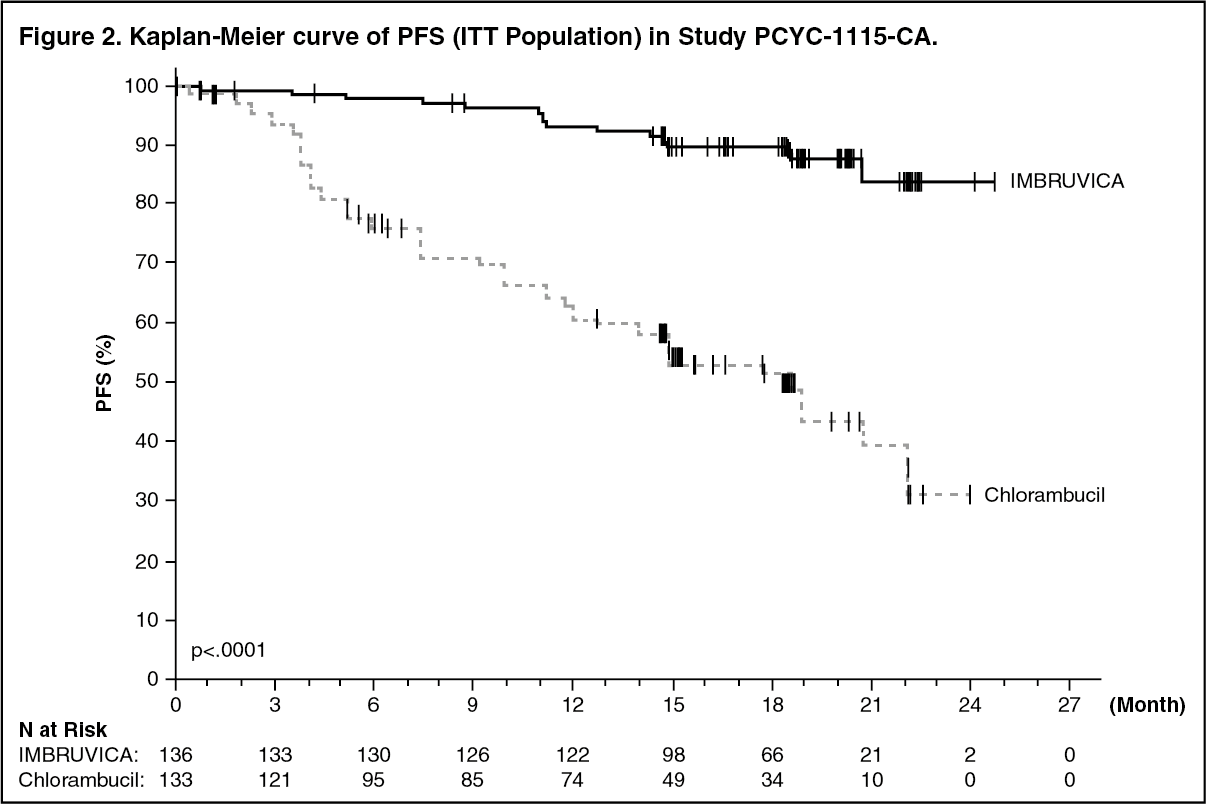 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image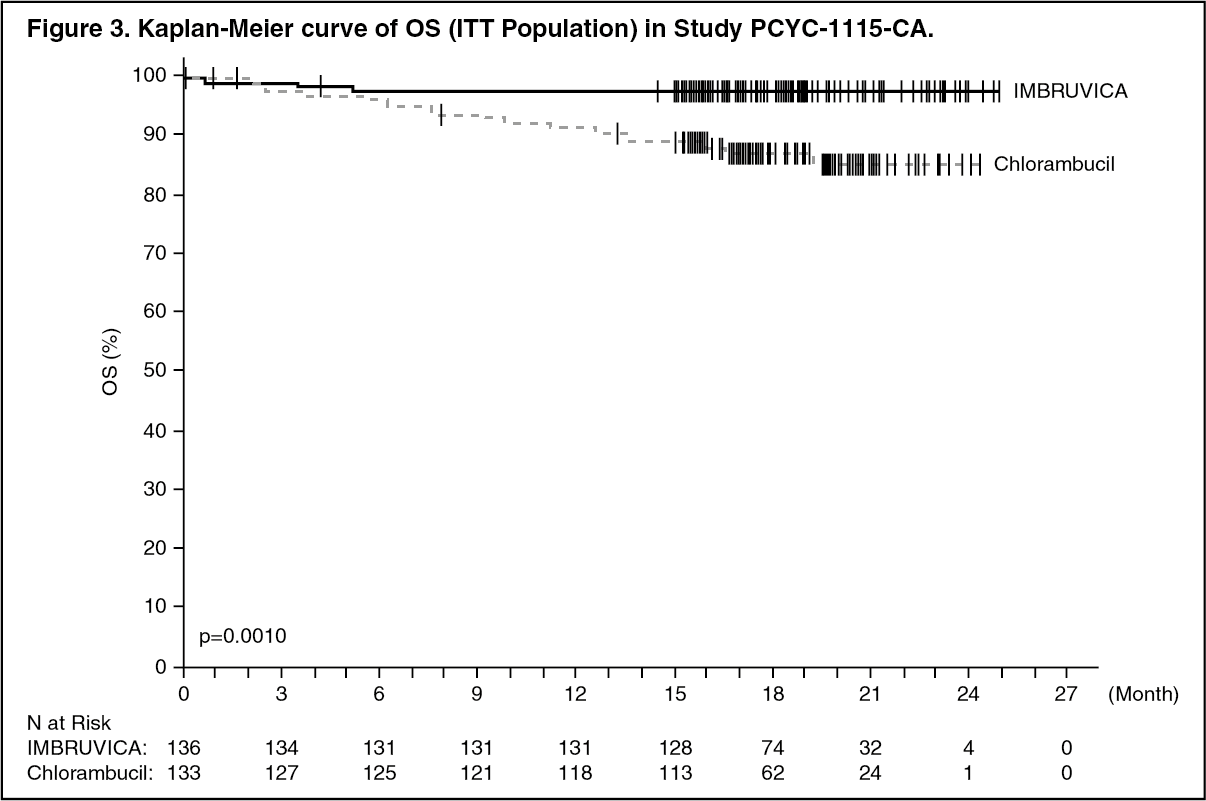 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image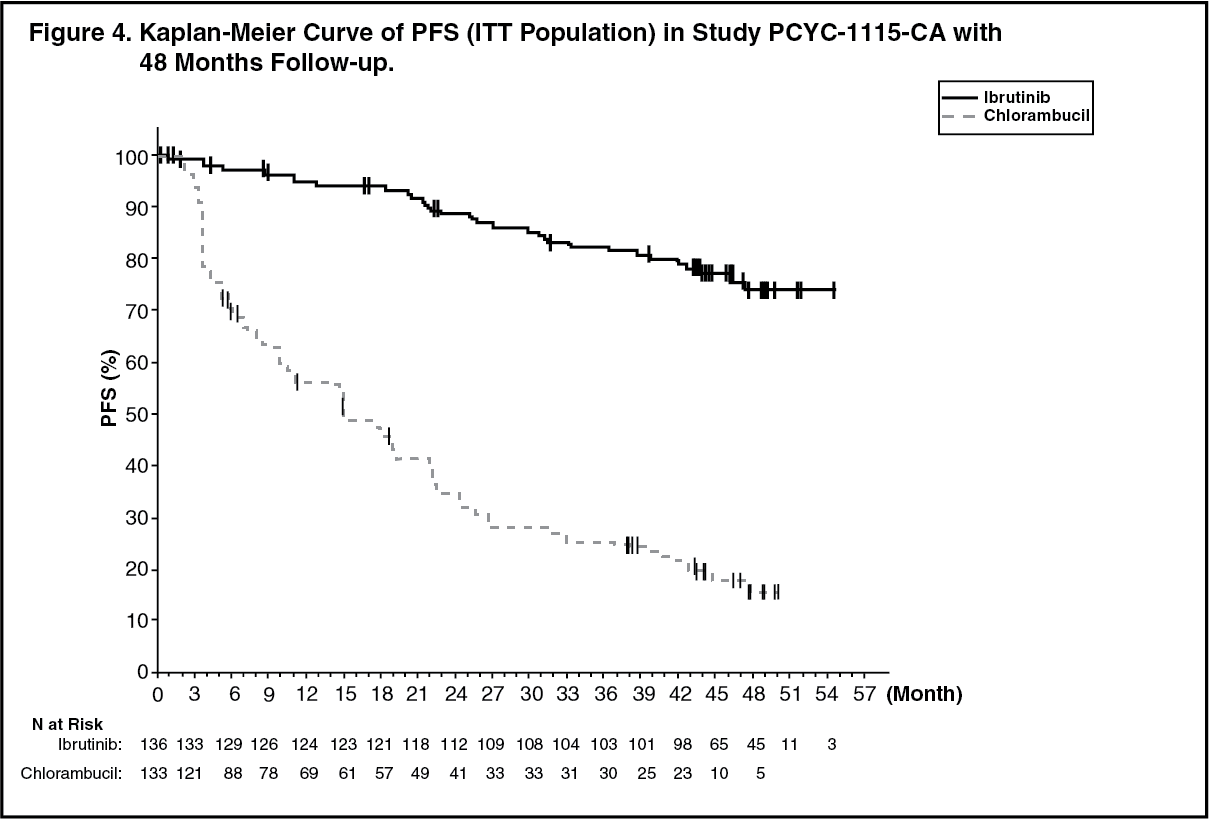 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image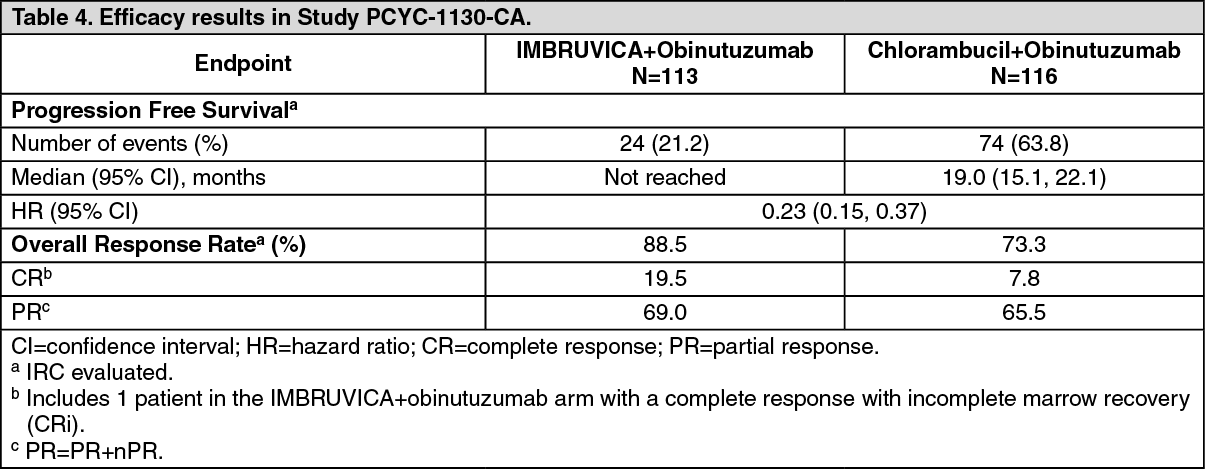 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image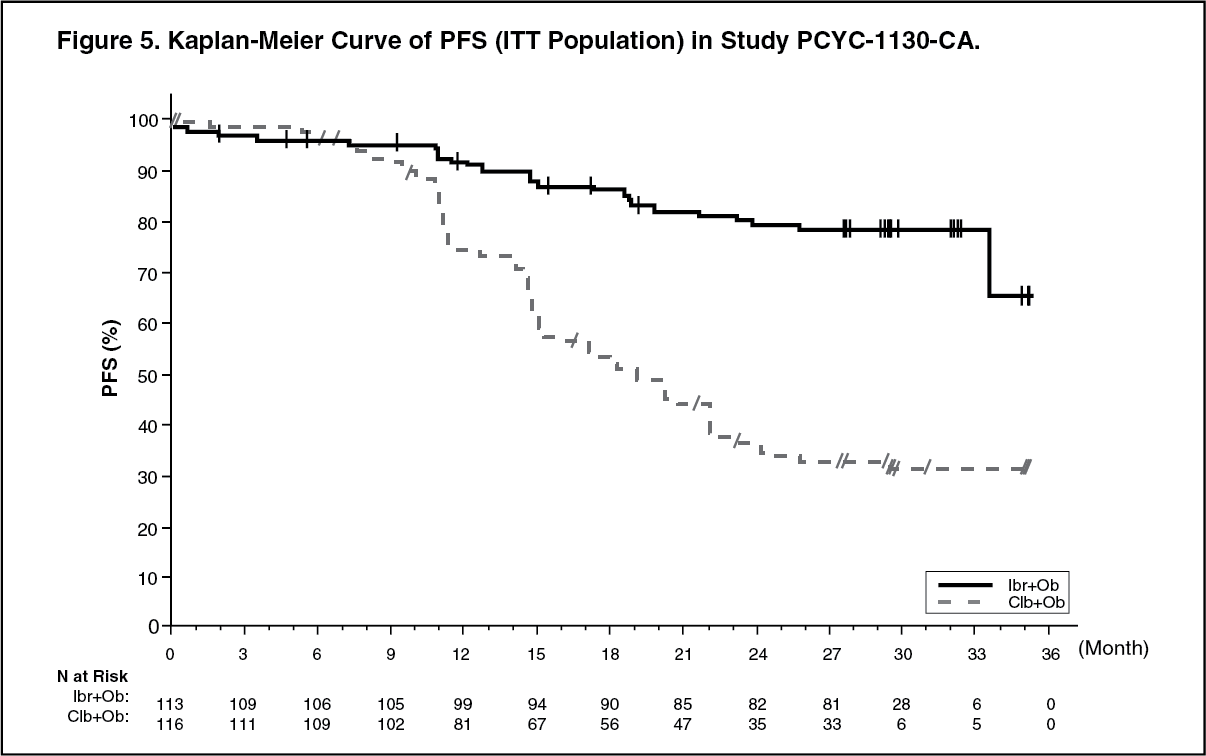 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image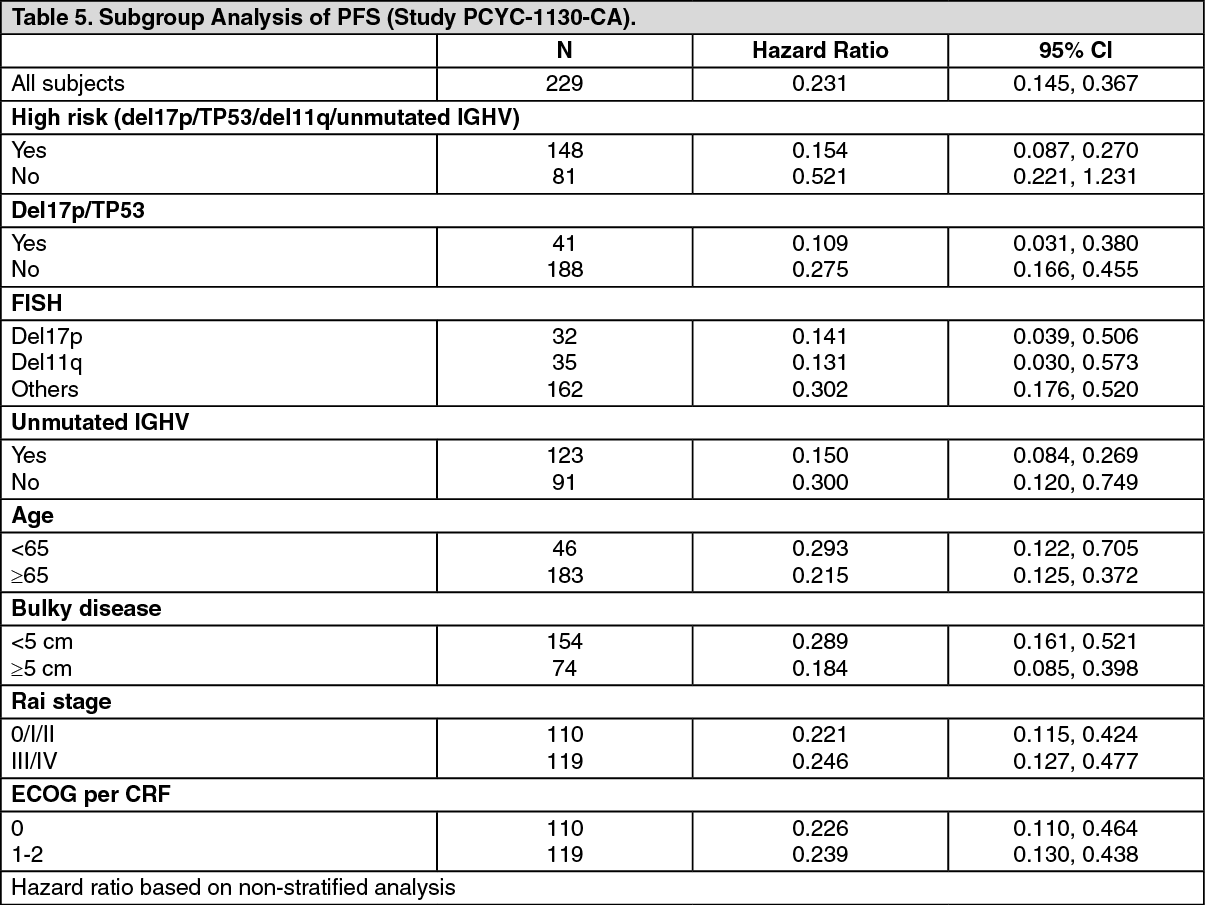 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image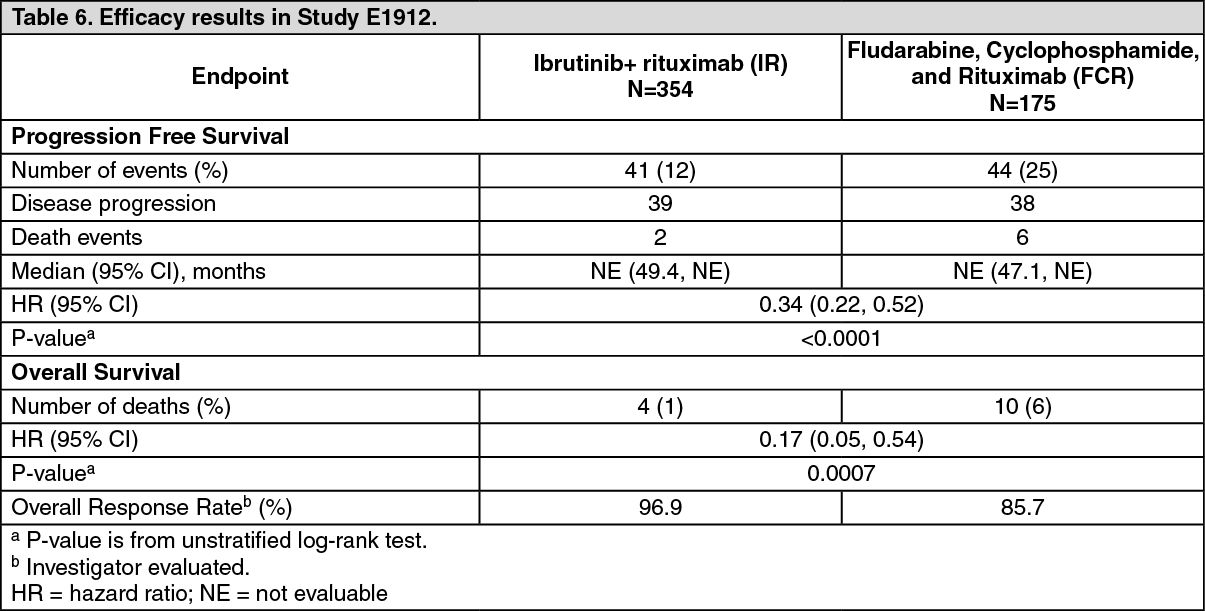 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image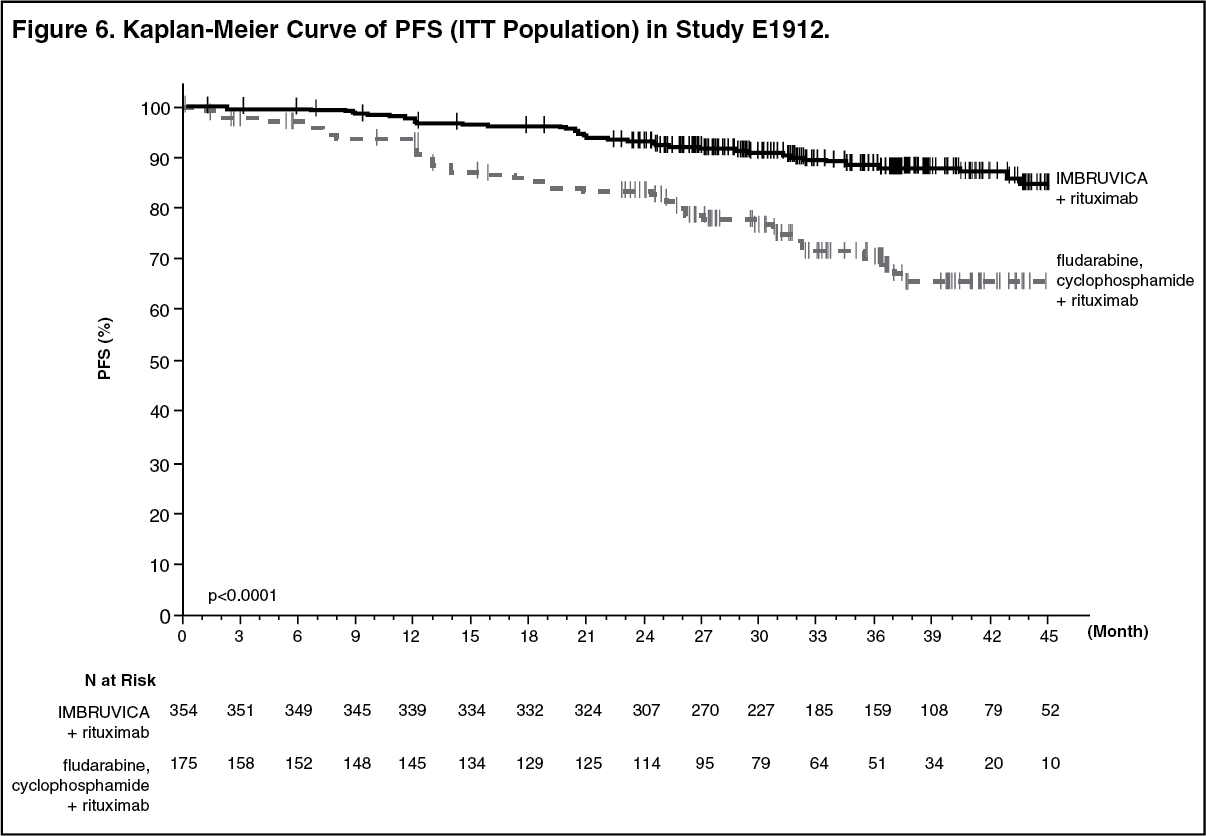 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image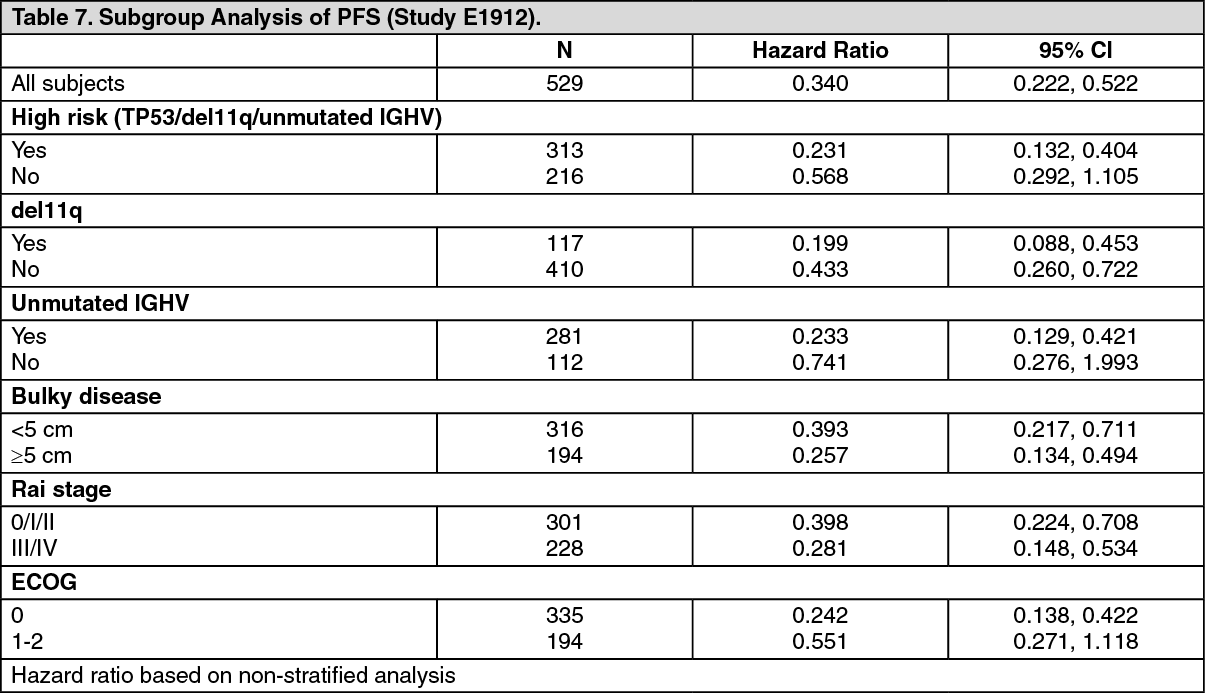 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image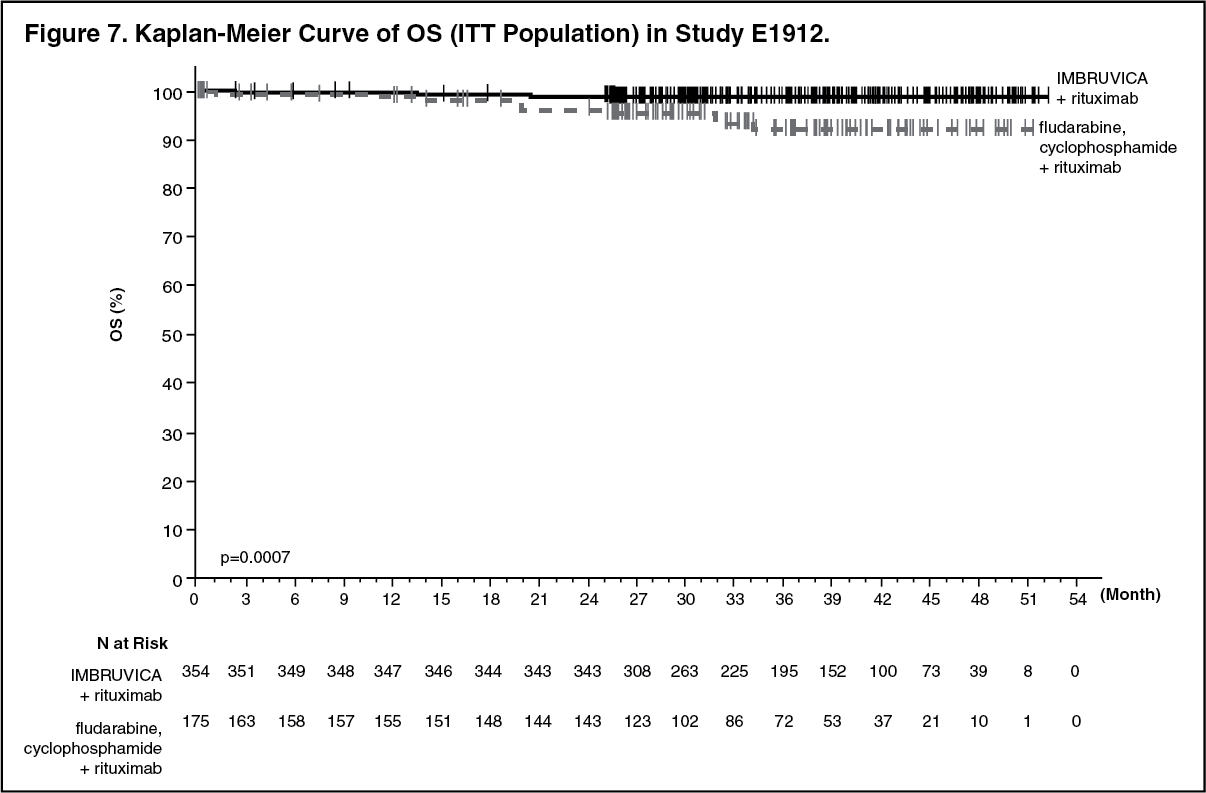 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image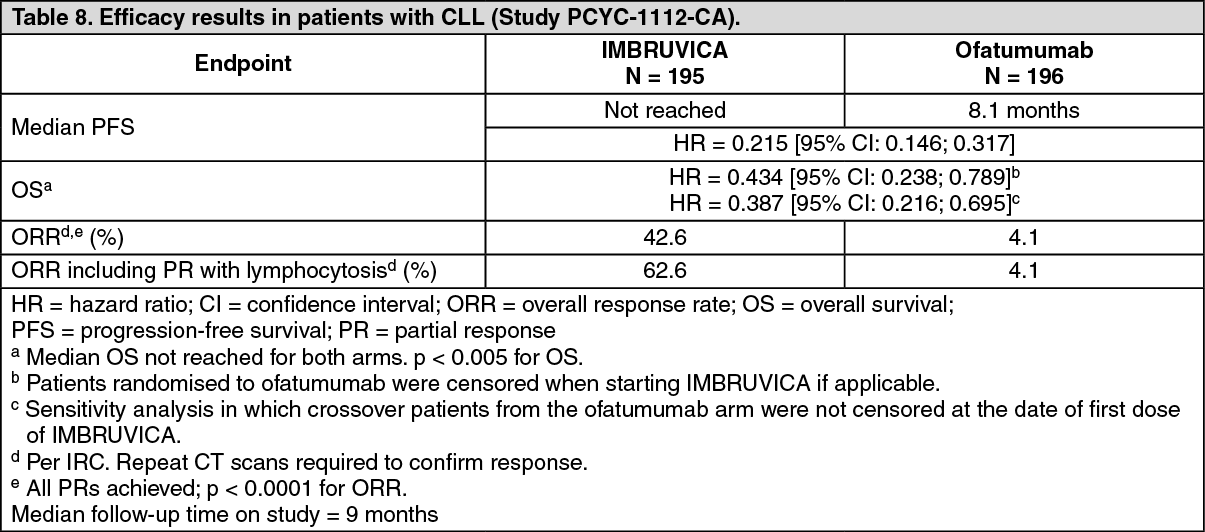 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image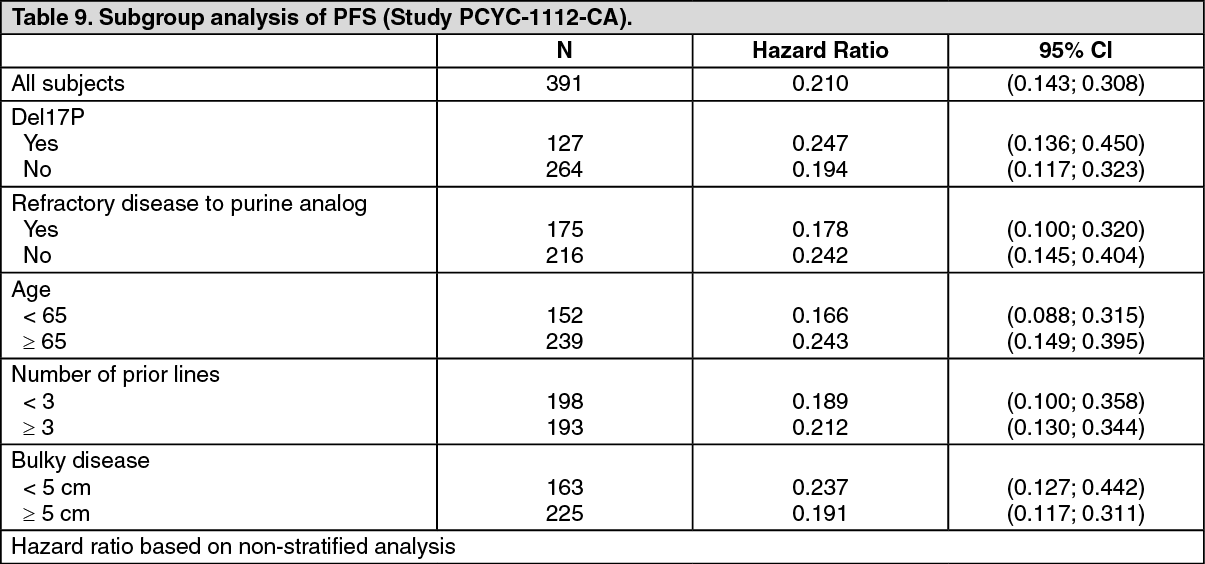 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image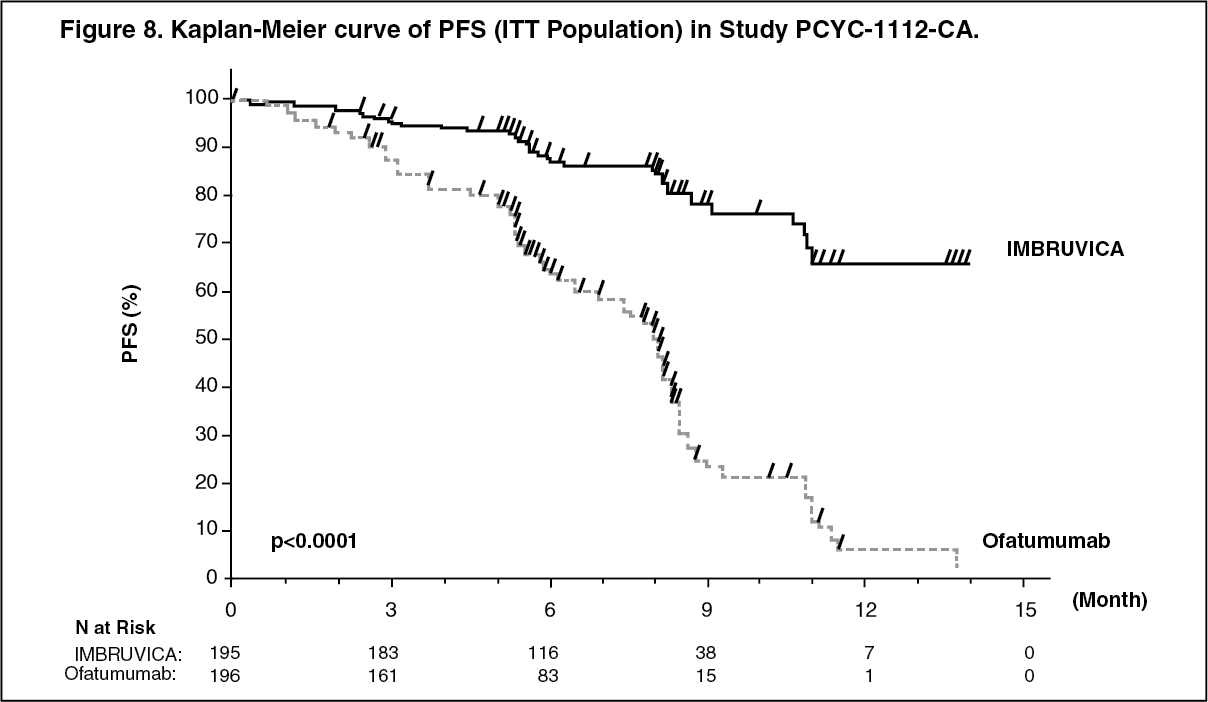 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image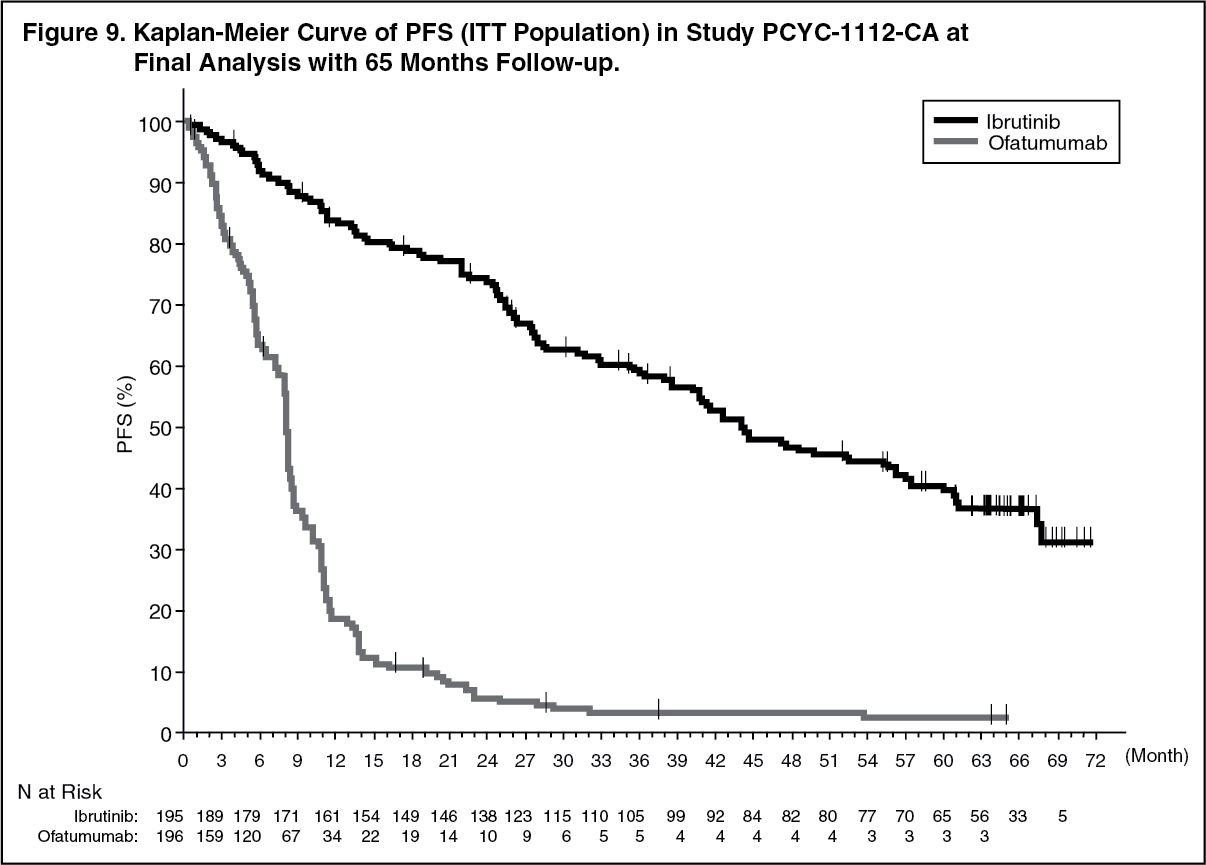 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image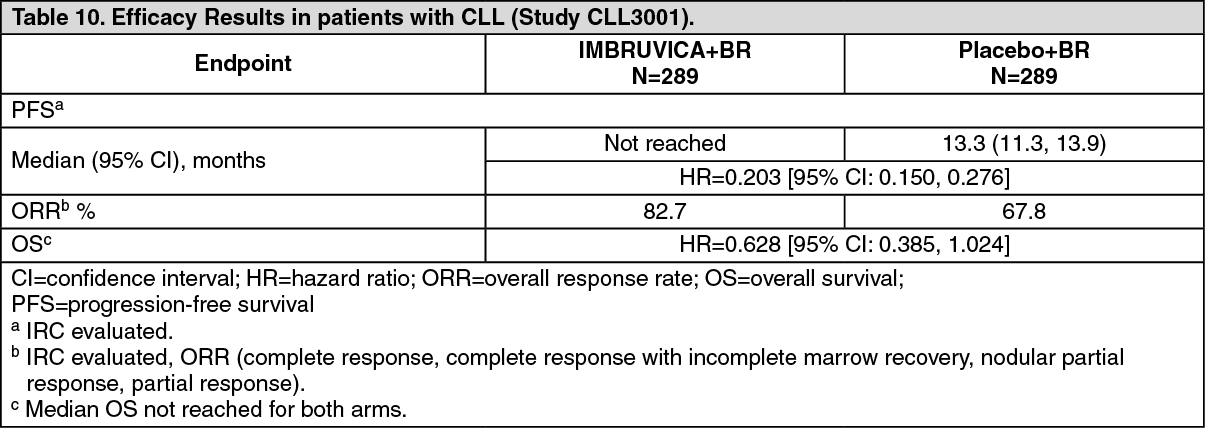 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image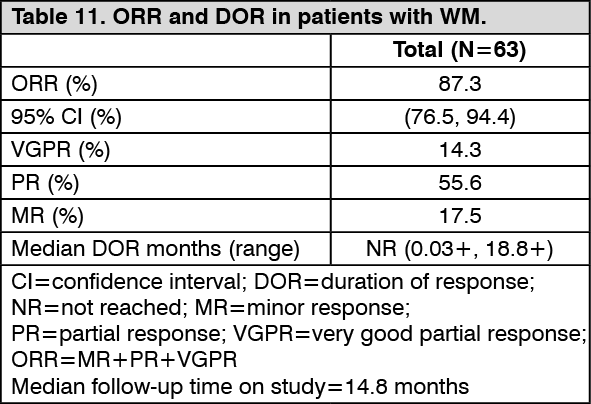 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image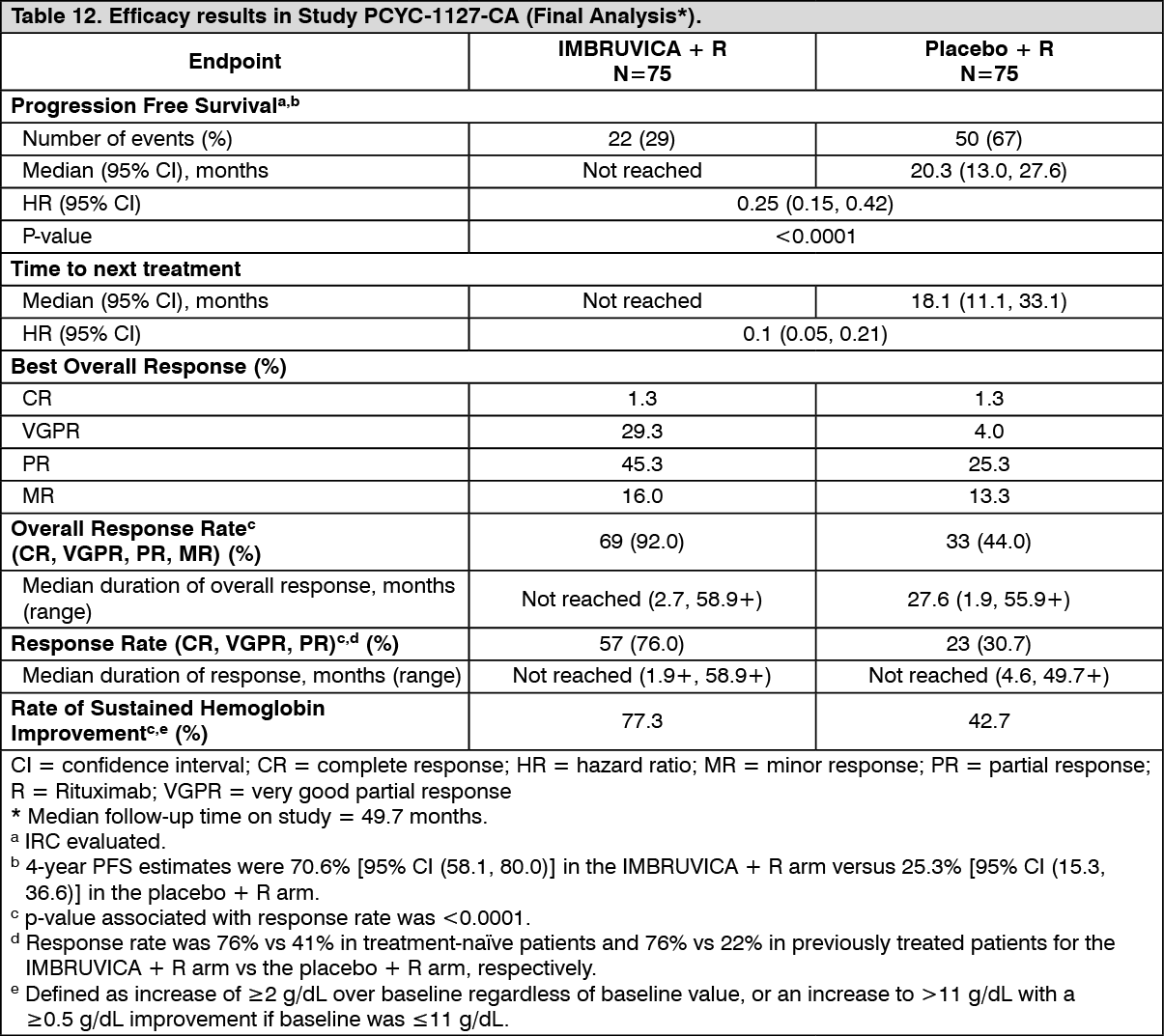 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image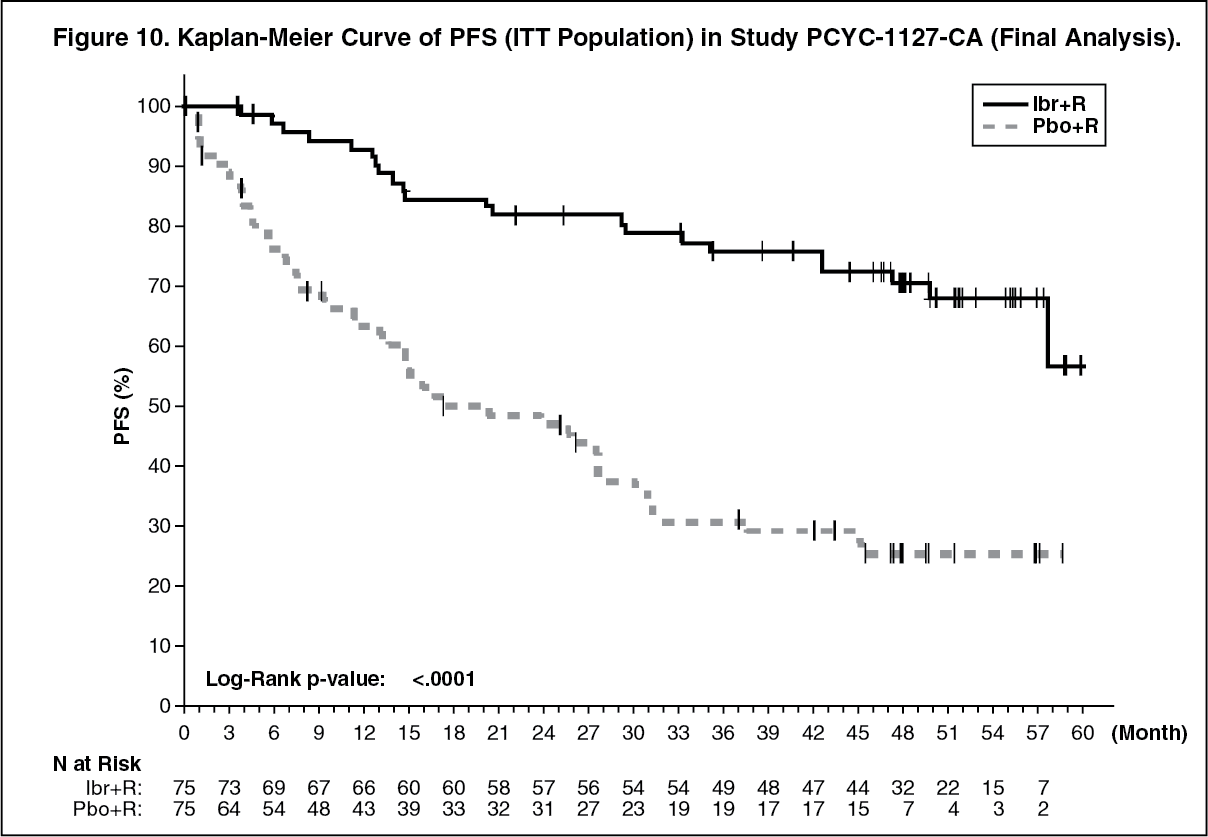 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image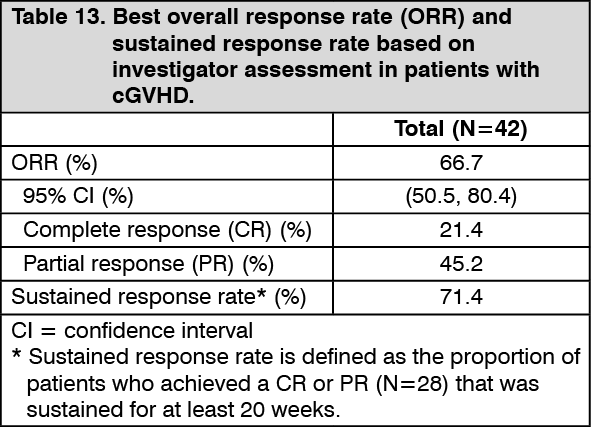 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image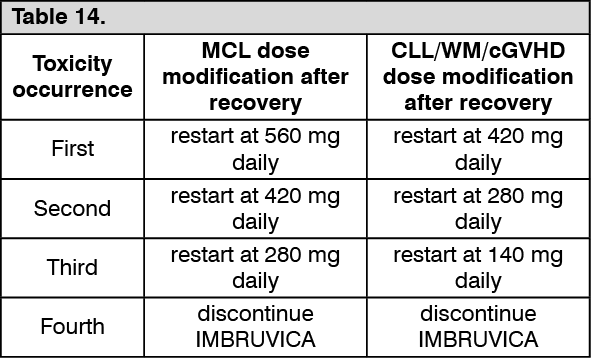 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image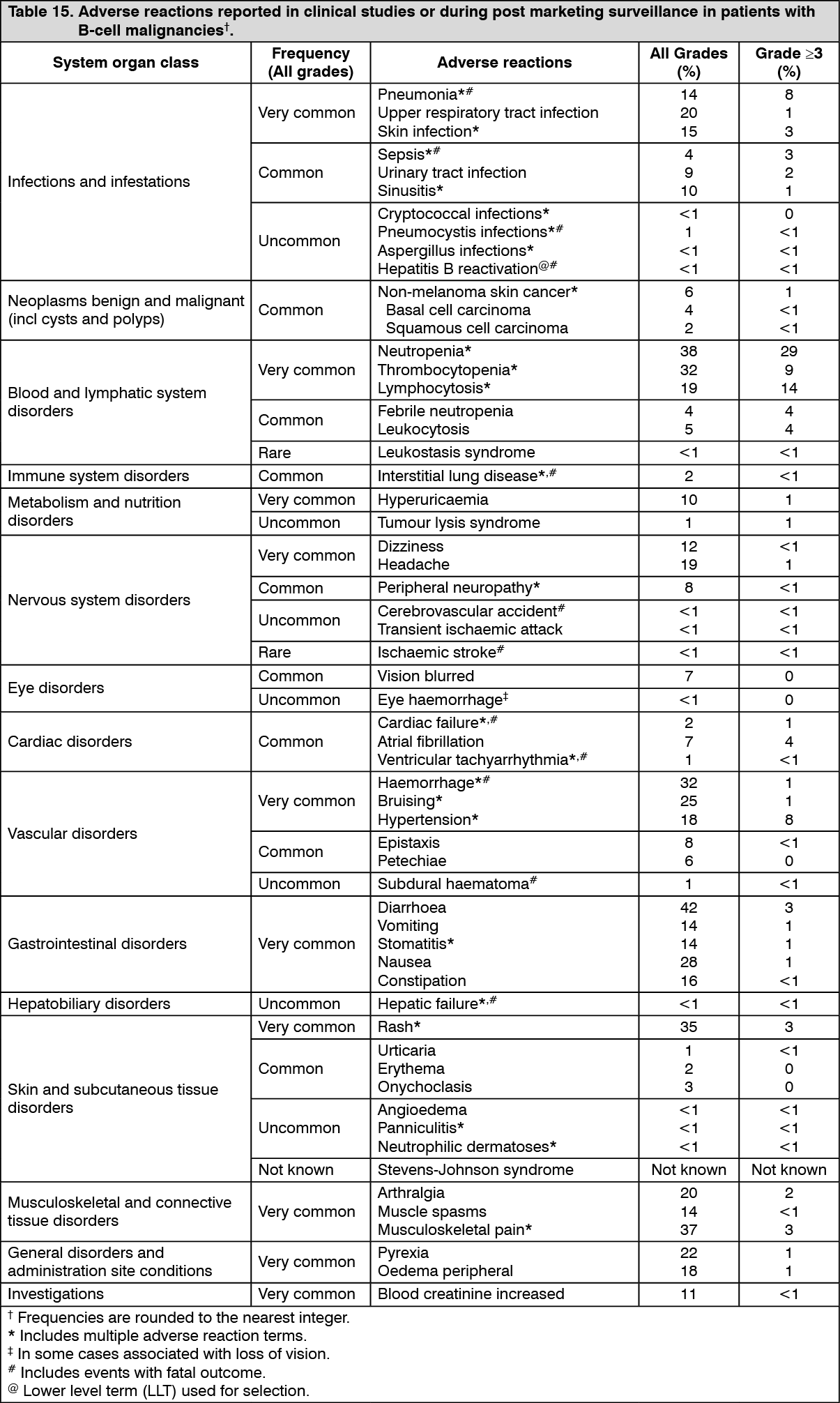 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image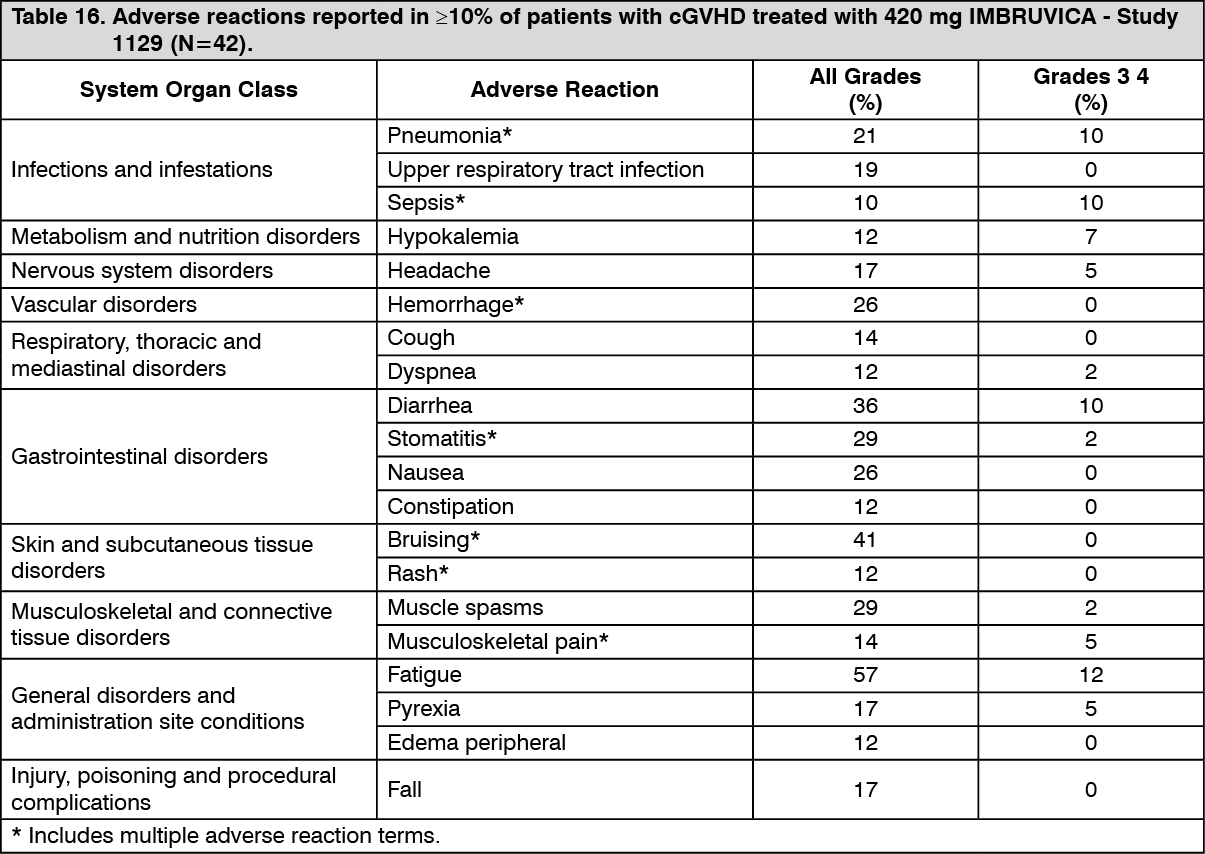 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image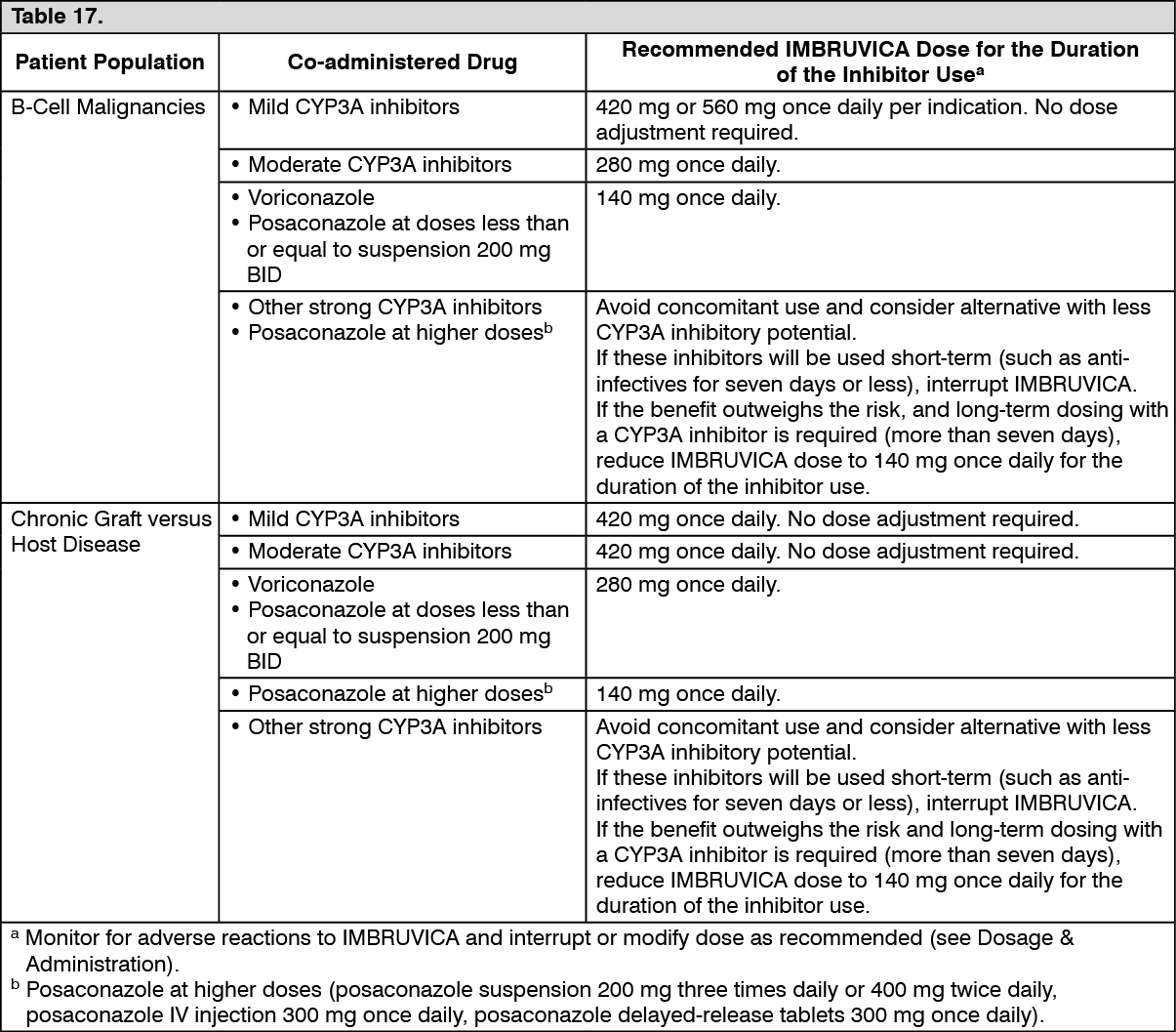 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
