


 Sign Out
Sign Out


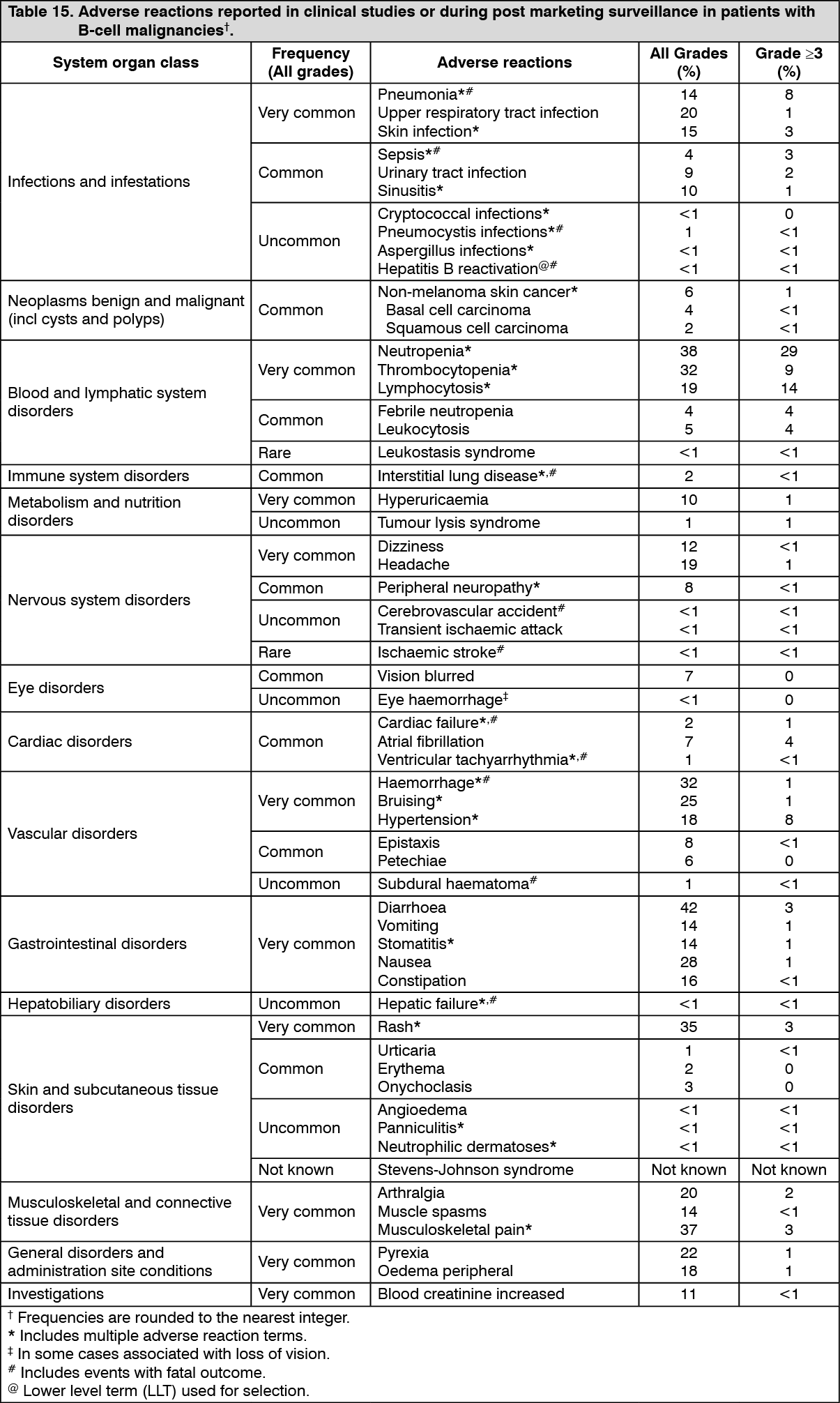
Tabulated list of adverse reactions: The safety profile is based on pooled data from 1552 patients treated with IMBRUVICA in three phase 2 clinical studies and seven randomised phase 3 studies and from post-marketing experience. Patients treated for MCL in clinical studies received IMBRUVICA at 560 mg once daily and patients treated for CLL or WM in clinical studies received IMBRUVICA at 420 mg once daily. All patients in clinical studies received IMBRUVICA until disease progression or no longer tolerated. The median duration of IMBRUVICA treatment across the pooled dataset was 17.4 months. The median duration of treatment for CLL/SLL was 18.2 months (up to 52 months); MCL was 11.7 months (up to 28 months); WM was 21.6 months (up to 37 months).
Adverse reactions in patients treated with ibrutinib for B-cell malignancies and post-marketing adverse reactions are listed as follows by system organ class and frequency grouping. Frequencies are defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), not known (cannot be estimated from the available data). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. (See Table 15.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Discontinuation and dose reduction due to adverse reactions: Of the 1552 patients treated with IMBRUVICA for B-cell malignancies, 6% discontinued treatment primarily due to adverse reactions. These included pneumonia, atrial fibrillation, thrombocytopenia, haemorrhage, neutropenia, rash, and athralgia. Adverse reactions leading to dose reduction occurred in approximately 8% of patients.
Elderly: Of the 1552 patients treated with IMBRUVICA, 52% were 65 years of age or older. Grade 3 or higher pneumonia (12% of patients age ≥65 versus 5% of patients <65 years) and thrombocytopenia (12% of patients age ≥65 years versus 6% of patients <65 years) occurred more frequently among elderly patients treated with IMBRUVICA.
Long-term safety: The safety data from long-term treatment with IMBRUVICA over 5 years from 1284 patients (treatment-naïve CLL/SLL n=162, relapsed/refractory CLL/SLL n=646, relapsed/refractory MCL n=370, and WM n=106) were analysed. The median duration of treatment for CLL/SLL was 51 months (range, 0.2 to 98 months) with 70% and 52% of patients receiving treatment for more than 2 years and 4 years, respectively. The median duration of treatment for MCL was 11 months (range, 0 to 87 months) with 31% and 17% of patients receiving treatment for more than 2 years and 4 years, respectively. The median duration of treatment for WM was 47 months (range, 0.3 to 61 months) with 78% and 46% of patients receiving treatment for more than 2 years and 4 years, respectively. The overall known safety profile of IMBRUVICA-exposed patients remained consistent, other than an increasing prevalence of hypertension, with no new safety concerns identified. The prevalence for Grade 3 or greater hypertension was 4% (year 0-1), 7% (year 1-2), 9% (year 2-3), 9% (year 3-4), and 9% (year 4-5); the overall incidence for the 5-year period was 11%.
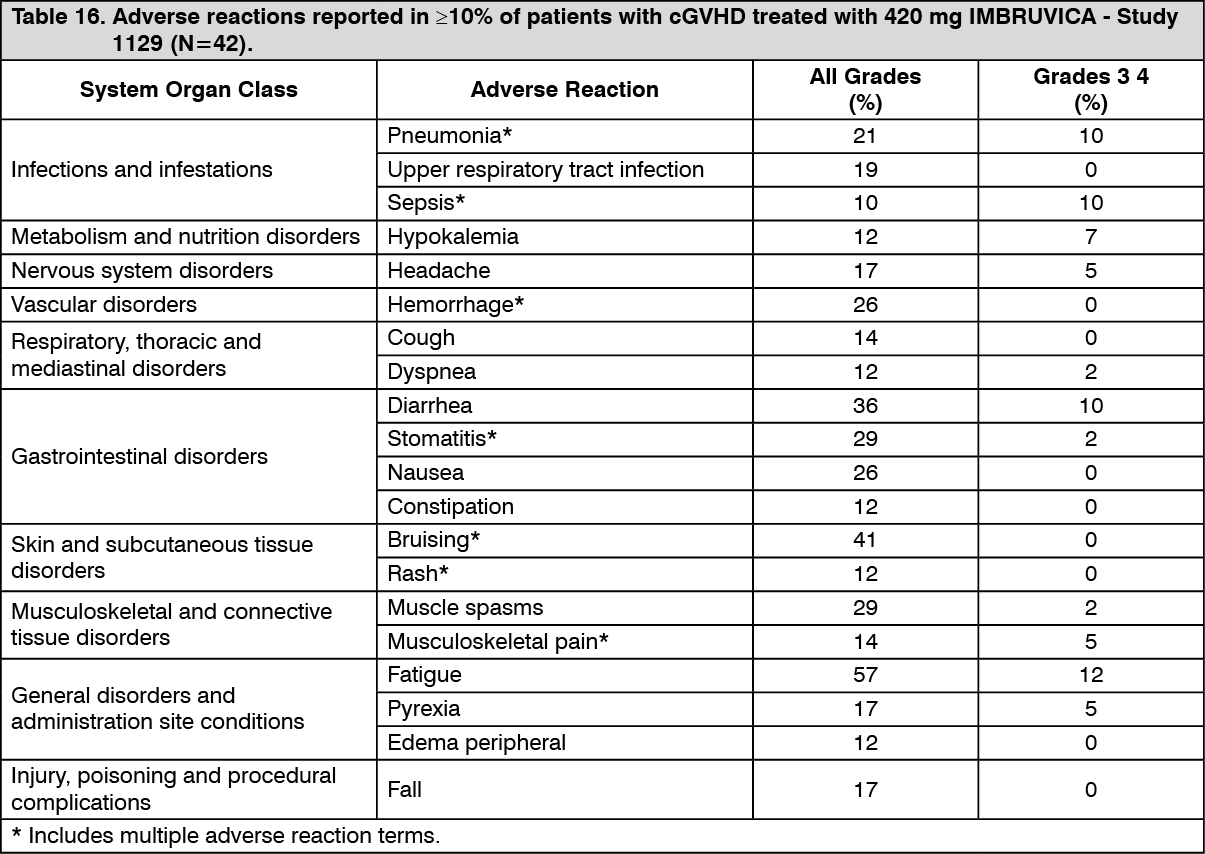
Chronic graft versus host disease: The data described as follows reflect exposure to IMBRUVICA in an open-label clinical study that included 42 patients with cGVHD after failure of first line corticosteroid therapy and required additional therapy.
The most commonly occurring adverse reactions in the cGVHD study (≥ 20%) were fatigue, bruising, diarrhea, stomatitis, muscle spasms, nausea, hemorrhage, and pneumonia. Atrial fibrillation occurred in one patient (2%), which was Grade 3.
Discontinuation and dose reduction due to ARs: Twenty-four percent of patients receiving IMBRUVICA in the cGVHD trial discontinued treatment due to adverse reactions. Adverse reactions leading to dose reduction occurred in 26% of patients.
Adverse reactions described as follows in Table 15 reflect exposure to IMBRUVICA with a median duration of 4.4 months in the cGVHD study. (See Table 16.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
View ADR Monitoring Form