Pharmacotherapeutic group: anti-anemic.
ATC code: B03XA01.
Pharmacology: Pharmacodynamics: Mechanism of action: Erythropoietin (EPO) is a glycoprotein hormone produced primarily by the kidney in response to hypoxia and is the key regulator of red blood cell (RBC) production. EPO is involved in all phases of erythroid development, and has its principal effect at the level of erythroid precursors. After EPO binds to its cell surface receptor, it activates signal transduction pathways that interfere with apoptosis and stimulates erythroid cell proliferation. Recombinant human EPO (Epoetin alfa), expressed in Chinese hamster ovary cells, has a 165 amino acid sequence identical to that of human urinary EPO; the 2 are indistinguishable on the basis of functional assays. The apparent molecular weight of erythropoietin is 32000 to 40000 dalton.
Pharmacodynamic effects: Pharmacodynamic responses to HSA-free Epoetin alfa, change in percent reticulocytes, hemoglobin, and total red blood cell counts as well as the area under the curve (AUCs) of these pharmacodynamic parameters, were similar between two dosing regimens (150 IU/kg SC three times per week to 40000 IU/mL SC once weekly).
ESAs are growth factors that primarily stimulate red cell production. Erythropoietin receptors may be expressed on the surface of a variety of tumor cells.
Healthy volunteers: After single doses (20000 to 160000 IU subcutaneously) of Epoetin alfa, a dose-dependent response was observed for the pharmacodynamic markers investigated including: reticulocytes, red blood cell count, and hemoglobin. A defined concentration-time profile with peak and return to baseline was observed for changes in percent reticulocytes. A less defined profile was observed for red blood cell count and hemoglobin. In general, all pharmacodynamic markers increased in a linear manner with dose reaching a maximum response at the highest dose levels.
Further pharmacodynamic studies explored 40000 IU once weekly versus 150 IU/kg 3 times per week. Despite differences in concentration-time profiles the pharmacodynamic response (as measured by changes in percent reticulocytes, hemoglobin, and total red blood cell count) was similar between these regimens. Additional studies compared the 40000 IU once-weekly regimen of Epoetin alfa with biweekly doses ranging from 80000 to 120000 IU subcutaneously. Overall, based on the results of these pharmacodynamic studies in healthy subjects, the 40000 IU once-weekly dosing regimen seems to be more efficient in producing red blood cells than the biweekly regimens despite an observed similarity in reticulocyte production in the once-weekly and biweekly regimens.
Chronic renal failure: Epoetin alfa has been shown to stimulate erythropoiesis in anemic subjects with CRF, including dialysis and pre-dialysis subjects. The first evidence of a response to Epoetin alfa is an increase in the reticulocyte count within 10 days, followed by increases in the red cell count, hemoglobin and hematocrit, usually within 2 to 6 weeks. The hemoglobin response varies between subjects and may be impacted by iron stores and the presence of concurrent medical problems.
Chemotherapy-induced anemia: Epoetin alfa administered 3 times per week or once weekly has been shown to increase hemoglobin and decrease transfusion requirements after the first month of therapy in anemic cancer subjects receiving chemotherapy.
In a study comparing the 150 IU/kg, 3 times-per-week and 40000 IU, once-weekly dosing regimens in healthy subjects and in anemic cancer subjects the time profiles of changes in percent reticulocytes, hemoglobin, and total red blood cells were similar between the two dosing regimens in both healthy and anemic cancer subjects. The AUCs of the respective pharmacodynamic parameters were similar between the 150 IU/kg, 3 times-per-week and 40000 IU, once-weekly dosing regimens in healthy subjects and also in anemic cancer subjects.
Adult surgery patients in an autologous predonation program: Epoetin alfa has been shown to stimulate red blood cell production in order to augment autologous blood collection, and to limit the decline in hemoglobin in adult patients scheduled for major elective surgery who are not expected to predeposit their complete perioperative blood needs.
Treatment of adult patients scheduled for major elective orthopedic surgery: In patients scheduled for major elective orthopedic surgery with a pretreatment hemoglobin of > 10 to ≤ 13 g/dL, Epoetin alfa has been shown to decrease the risk of receiving allogeneic transfusions and hasten erythroid recovery (increased hemoglobin levels, hematocrit levels, and reticulocyte counts).
Clinical studies: Chronic renal failure: Epoetin alfa has been studied in clinical trials in adult anemic CRF patients, including patients on dialysis and patients not yet on dialysis, to treat anemia and maintain hematocrit within a concentration range of 30-36%.
In clinical trials at starting doses of 50-150 IU/kg three times per week, approximately 95% of all patients responded with a clinically significant increase in hematocrit. After approximately two months of therapy, virtually all patients were transfusion-independent. Once the hematocrit concentration range was achieved, the maintenance dose was individualized for each patient. In the three largest clinical trials conducted in adult patients on dialysis, the median maintenance dose necessary to maintain the hematocrit between 30-36% was approximately 75 IU/kg given three times per week.
In a double-blind, placebo-controlled, multicenter, quality of life study in CRF patients on hemodialysis, clinically and statistically significant improvement was shown in the patients treated with Epoetin alfa compared to the placebo group when measuring fatigue, physical symptoms, relationships and depression (Kidney Disease Questionnaire) after six months of therapy. Patients from the group treated with Epoetin alfa were also enrolled in an open-label extension study which demonstrated improvements in their quality of life were maintained for an additional 12 months.
Adult patients with renal insufficiency not yet undergoing dialysis: In clinical trials conducted in patients with CRF not on dialysis treated with Epoetin alfa, the average duration of therapy was nearly five months. These patients responded to Epoetin alfa therapy in a manner similar to that observed in patients on dialysis. Patients with CRF not on dialysis demonstrated a dose-dependent and sustained increase in hematocrit when Epoetin alfa was administered by either an intravenous or subcutaneous route. Similar rates of rise of hematocrit were noted when Epoetin alfa was administered by either route. Moreover, Epoetin alfa doses of 75-150 IU/kg per week have been shown to maintain hematocrits of 36-38% for up to six months.
A randomized prospective trial (CHOIR) evaluated 1432 anemic chronic renal failure patients who were not undergoing dialysis. Patients were assigned to Epoetin alfa treatment targeting a maintenance hemoglobin level of 13.5 g/dL (higher than the recommended target hemoglobin level) or 11.3 g/dL. A major cardiovascular event (death, myocardial infarction, stroke or hospitalization for congestive heart failure) occurred among 125 (18%) of the 715 patients in the higher hemoglobin group compared to 97 (14%) among the 717 patients in the lower hemoglobin group (hazard ratio [HR] 1.3, 95% CI: 1.0, 1.7, p = 0.03).
Treatment of patients with chemotherapy induced anemia: Epoetin alfa has been studied in clinical trials in adult anemic cancer patients with lymphoid and solid tumors, and patients on various chemotherapy regimens, including platinum and non-platinum-containing regimens. In these trials, Epoetin alfa administered three times a week (tiw) and once weekly has been shown to increase hemoglobin and decrease transfusion requirements after the first month of therapy in anemic cancer patients. In some studies, the double-blind phase was followed by an open-label phase during which all patients received Epoetin alfa and a maintenance of effect was observed.
Available evidence suggests the hematopoietic response to Epoetin alfa therapy is similar between patients with non-myeloid hematologic and solid tumors and in patients with or without tumor bone marrow infiltration. Comparable intensity of chemotherapy in the Epoetin alfa and placebo groups in the chemotherapy trials was demonstrated by a similar area under the neutrophil time curve in patients treated with Epoetin alfa and placebo-treated patients, as well as by a similar proportion of patients in groups treated with Epoetin alfa and placebo-treated groups whose absolute neutrophil counts fell below 1000 and 500 cells/mcL.
In a prospective, randomized, double-blind, placebo-controlled trial conducted in 375 anemic patients with various non-myeloid malignancies receiving non-platinum chemotherapy, there was a significant reduction of anemia-related sequelae (e.g. fatigue, decreased energy, and activity reduction), as measured by the following instruments and scales: Functional Assessment of Cancer Therapy-Anemia (FACT-An) general scale, FACT-An fatigue scale, and Cancer Linear Analogue Scale (CLAS).
A randomized, open-label, multicenter study was conducted in 2098 anemic women with metastatic breast cancer, who received first line or second line chemotherapy. This was a non- inferiority study designed to rule out a 15% risk increase in tumor progression or death of epoetin alfa plus SOC as compared with SOC alone. The median progression free survival (PFS) per investigator assessment of disease progression was 7.4 months in each arm (HR 1.09, 95% CI: 0.99, 1.20), indicating the study objective was not met. Median PFS with disease progression assessed by the Independent Review Committee was 7.6 months in each arm (HR 1.03, 95% CI: 0.92, 1.15). At clinical cutoff, 1337 deaths were reported. Median overall survival in the epoetin alfa plus SOC group was 17.2 months compared with 17.4 months in the SOC alone group (HR 1.06, 95% CI: 0.95, 1.18). Significantly fewer patients received RBC transfusions in the epoetin alfa plus SOC arm (5.8% versus 11.4%); however, significantly more patients had TVEs in the epoetin alfa plus SOC arm (2.8% versus 1.4%). At the final analysis, 1653 deaths were reported. Median overall survival in the epoetin alfa plus SOC group was 17.8 months compared with 18.0 months in the SOC alone group (HR 1.07, 95% CI: 0.97, 1.18). The median time to progression (TTP) based on investigator-determined progressive disease (PD) was 7.5 months in the epoetin alfa plus SOC group and 7.5 months in the SOC group (HR 1.099, 95% CI: 0.998, 1.210). The median TTP based on IRC-determined PD was 8.0 months in the epoetin alfa plus SOC group and 8.3 months in the SOC group (HR 1.033, 95% CI: 0.924, 1.156).
The totality of evidence, including results of meta-analyses and clinical experience from controlled studies of ESAs in patients with cancer, continues to support a favorable benefit-risk balance for the use of ESAs in patients with chemotherapy-induced anemia, when used according to the prescribing information. In meta-analyses of studies in which patients were receiving chemotherapy there were no statistically significant increases in either mortality or tumor progression. Signals in individual studies conducted outside of the recommendations in the product labeling (hemoglobin targets above 12 g/dL and/or no chemotherapy treatment) have raised concerns (see Precautions, Cancer Patients).
Autologous predonation program: The effect of Epoetin alfa in facilitating autologous blood donation in patients with low hematocrits (≤ 39% and no underlying anemia due to iron deficiency) scheduled for major orthopedic surgery was evaluated in a double-blind, placebo-controlled study conducted in 204 subjects, and a single-blind placebo controlled study in 55 subjects.
In the double-blind study, subjects were treated with Epoetin alfa 600 IU/kg or placebo intravenously once daily every 3 to 4 days over 3 weeks (total 6 doses). On average, subjects treated with Epoetin alfa were able to predeposit significantly more units of blood (4.5 units) than placebo-treated subjects (3.0 units).
In a study where the subject, surgeon and anesthesiologist were blinded, subjects were treated with Epoetin alfa 300 IU/kg or 600 IU/kg or placebo intravenously once daily every 3 to 4 days over 3 weeks (total 6 doses). Subjects treated with Epoetin alfa were also able to predeposit significantly more units of blood (Epoetin alfa 300 IU/kg = 4.4 units; Epoetin alfa 600 IU/kg = 4.7 units) than placebo-treated subjects (2.9 units).
Epoetin alfa therapy reduced the risk of exposure to allogeneic blood by 50% compared to subjects not receiving Epoetin alfa.
Major elective orthopedic surgery: The effect of Epoetin alfa (300 IU/kg or 100 IU/kg) on the exposure to allogeneic blood transfusion has been evaluated in a placebo-controlled, double-blind clinical trial in non-iron deficient adult subjects scheduled for major elective orthopedic hip or knee surgery. Epoetin alfa was administered subcutaneously for 10 days prior to surgery, on the day of surgery, and for four days after surgery. Subjects were stratified according to their baseline hemoglobin (≤ 10 g/dL, > 10 to ≤ 13 g/dL and > 13 g/dL).
Epoetin alfa 300 IU/kg significantly reduced the risk of allogeneic transfusion in subjects with a pretreatment hemoglobin of > 10 to ≤ 13 g/dL. Sixteen percent of Epoetin alfa 300 IU/kg, 23% of Epoetin alfa 100 IU/kg and 45% of placebo-treated subjects required transfusion.
An open-label, parallel-group trial in non-iron deficient adult subjects with a pretreatment hemoglobin of ≥ 10 to ≤ 13 g/dL who were scheduled for major orthopedic hip or knee surgery compared Epoetin alfa 300 IU/kg subcutaneously daily for 10 days prior to surgery, on the day of surgery and for four days after surgery to Epoetin alfa 600 IU/kg subcutaneously once weekly for 3 weeks prior to surgery and on the day of surgery.
From pretreatment to presurgery, the mean increase in hemoglobin in the 600 IU/kg weekly group (1.44 g/dL) was twice than that observed in the 300 IU/kg daily group (0.73 g/dL). Mean hemoglobin levels were similar for the two treatment groups throughout the postsurgical period. The erythropoietic response observed in both treatment groups resulted in similar transfusion rates (16% in the 600 IU/kg weekly group and 20% in the 300 IU/kg daily group).
Adult patients with low- or intermediate-1-risk MDS: A randomized, double-blind, placebo-controlled, multicenter study evaluated the efficacy and safety of epoetin alfa in adult anemic subjects with low- or intermediate-1-risk MDS.
Erythroid response was defined according to IWG 2006 criteria as a hemoglobin increase ≥ 1.5 g/dL from baseline or a reduction of RBC units transfused by an absolute number of at least 4 units every 8 weeks compared to the 8 weeks prior to baseline, and a response duration of at least 8 weeks.
Erythroid response during the first 24 weeks of the study was demonstrated by 27/85 (31.8%) of the subjects in the epoetin alfa group compared to 2/45 (4.4%) of the subjects in the placebo group (p<0.001).
Median time from baseline to first transfusion was statistically significantly longer in the epoetin alfa group compared to placebo (49 vs. 37 days; p=0.046). After 4 weeks of treatment the time to first transfusion was further increased in the epoetin alfa group (142 vs. 50 days, p=0.007). The percentage of subjects who were transfused in the epoetin alfa group decreased from 51.8% in the 8 weeks prior to baseline to 24.7% between weeks 16 and 24, compared to the placebo group which had an increase in transfusion rate from 48.9% to 54.1% over the same time periods.
Pediatric Population: Chronic renal failure: Epoetin alfa was evaluated in an open-label, non-randomized, escalating dosing, 52-week clinical study in pediatric CRF subjects undergoing hemodialysis. The median age of subjects enrolled in the study was 11.6 years (range 0.5 to 20.1 years).
Epoetin alfa was administered at 75 IU/kg/week intravenously in 2 or 3 divided doses post-dialysis, titrated by 75 IU/kg/week at intervals of 4 weeks (up to a maximum of 300 IU/kg/week), to achieve a 1 g/dL/month increase in hemoglobin. The desired hemoglobin concentration range was 9.6 to 11.2 g/dL. Eighty-one percent of subjects achieved hemoglobin concentrations in the desired range. The median time to target was 11 weeks and the median dose at target was 150 IU/kg/week. Of the subjects who achieved the target, 90% did so on a 3 times-per-week dosing regimen.
After 52 weeks, 57% of subjects remained in the study, receiving a median dose of 200 IU/kg/week.
Clinical data with subcutaneous administration in children are limited. In 5 small, open label, uncontrolled studies (number of patients ranged from 9-22, total N=72), epoetin alfa was administered subcutaneously in children at starting doses of 100 IU/kg/week to 150 IU/kg/week with the possibility to increase up to 300 IU/kg/week. In these studies, most were predialysis patients (N=44), 27 patients were on peritoneal dialysis and 2 were on hemodialysis with age ranging from 4 months to 17 years. Overall, these studies have methodological limitations but treatment was associated with positive trends towards higher hemoglobin levels. No unexpected adverse events were reported.
Pharmacokinetics: Intravenous Administration: Measurement of Epoetin alfa following multiple dose intravenous (IV) administration of 50 to 100 IU/kg revealed a half-life of approximately 4 hours in healthy subjects and a longer half-life in renal failure patients of approximately 5 hours after doses of 50, 100, and 150 IU/kg. A half-life of approximately 6 hours has been reported in children. With at least 4 days of PK blood sampling, half-life estimates ranging from 20.1 to 33.0 hours were observed in cancer subjects receiving 667 and 1500 IU/kg IV doses of Epoetin alfa.
Subcutaneous Administration: Serum concentrations following subcutaneous injection are lower than those following intravenous injection. Serum levels increase slowly and reach a peak 12 to 18 hours after subcutaneous dosing. The peak serum concentration is below the peak observed using the intravenous route (approximately 1/20
th of the value).
There is no accumulation: serum levels remain the same, whether data are collected 24 hours after the first injection or 24 hours after the last injection. Concentration-time profiles of erythropoietin on Week 1 and Week 4 were similar during multiple dosing of 600 IU/kg/once weekly in healthy subjects.
The pharmacokinetic data indicate no apparent difference in half-life among adult patients above or below 65 years of age.
A study of 7 preterm very low birth weight neonates and 10 healthy adults given IV erythropoietin suggested that distribution volume was approximately 1.5 to 2 times higher in the preterm neonates than in the healthy adults, and clearance was approximately 3 times higher in the preterm neonates than in the healthy adults.
The half-life for the subcutaneous route of administration is approximately 24 hours. Mean half-life values in healthy subjects were 19.4 ± 8.1 and 15.0 ± 6.1 with multiple dosing of 150 IU/kg three times per week and 40,000 IU/mL once weekly, respectively.
In a study comparing 40,000 IU SC once weekly to 150 IU/kg SC three times per week dosing regimens of HSA-containing Epoetin alfa in healthy subjects, the following parameters were estimated using data corrected for predose endogenous erythropoietin concentration during Week 4: (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Based on AUC comparison, relative bioavailability of Epoetin alfa 40,000 IU once weekly versus 150 IU/kg three times per week was 176%.
In a study comparing 40,000 IU SC once weekly versus 150 IU/kg SC three times per week dosing of HSA-free Epoetin alfa in healthy subjects, the following parameters were estimated using data corrected for predose endogenous erythropoietin concentration during Week 4: (See Table 3.)
Click on icon to see table/diagram/image
Based on AUC comparison, relative bioavailability of Epoetin alfa 40,000 IU/mL once weekly versus 150 IU/kg three times per week was 239%.
The bioavailability of subcutaneous Epoetin alfa after a dose of 120 IU/kg is much lower than that of the intravenous drug: approximately 20%.
Pharmacokinetic parameters were estimated in healthy subjects and anemic cancer subjects receiving cyclic chemotherapy and either 150 IU/kg three times per week or 40,000 IU/mL once weekly of HSA-containing Epoetin alfa. The pharmacokinetic parameters of anemic cancer subjects were different from those observed in healthy subjects during Week 1 (when the anemic cancer subjects were receiving chemotherapy) but were similar during Week 3 (when the anemic cancer subjects were not receiving chemotherapy). (See Table 4.)
Click on icon to see table/diagram/image
Pharmacokinetics of HSA-free Epoetin alfa were studied in anemic cancer subjects receiving cyclic chemotherapy after the 150 IU/kg three times per week and 40000 IU/mL once weekly dosing regimens. In general, there was a high degree of variability associated with the pharmacokinetic parameters in anemic cancer subjects. In general, the first pharmacokinetic profile of Epoetin alfa during Week 1 (when the anemic cancer subjects were receiving chemotherapy) demonstrated higher C
max, increased half-life, and lower clearance than the second pharmacokinetic profile during Week 3 or 4 (when the anemic cancer subjects were not receiving chemotherapy). (See Table 5.)
Click on icon to see table/diagram/image
Toxicology: NON-CLINICAL INFORMATION: Chronic Toxicity: In repeated dose toxicological studies in dogs and rats, but not in monkeys, Epoetin alfa therapy was associated with subclinical bone marrow fibrosis. Bone marrow fibrosis is a known complication of chronic renal failure in humans; it may be related to secondary hyperparathyroidism or unknown factors. In one study, there was no difference in the incidence of bone marrow fibrosis in hemodialysis patients treated with Epoetin alfa for 3 years and hemodialysis patients not treated with Epoetin alfa.
Carcinogenicity and Mutagenicity: Carcinogenicity: Long-term carcinogenicity studies have not been carried out. There are conflicting reports in the literature regarding ESAs as tumor proliferators. The clinical significance of these reports, based on
in vitro findings from human tumor samples, is unknown.
Mutagenicity: Epoetin alfa does not induce bacterial gene mutation (Ames Test), chromosomal aberrations in mammalian cells, micronuclei in mice, or gene mutation at the HGPRT locus.
Reproductive Toxicology: Preclinical studies have shown no evidence of teratogenicity in rats or rabbits at dosages up to 500 IU/kg/day administered intravenously. However, intravenous administration of Epoetin alfa causes a slight but not statistically significant decrease in fertility at 500 IU/kg, increased pre- and post-implantation loss and decreased fetal body weight at 100 and 500 IU/kg/day and delayed ossification at 20, 100, and 500 IU/kg/day. The latter finding was associated with reduced maternal body weight. Intravenous administration to lactating rats resulted in decreases in body weight gain, delays in appearance of abdominal hair and eyelid opening, and decreases in the number of caudal vertebra in the F
1 fetuses of the 500 IU/kg/day group. There were no Epoetin alfa-related effects on the F
2 generation fetuses.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image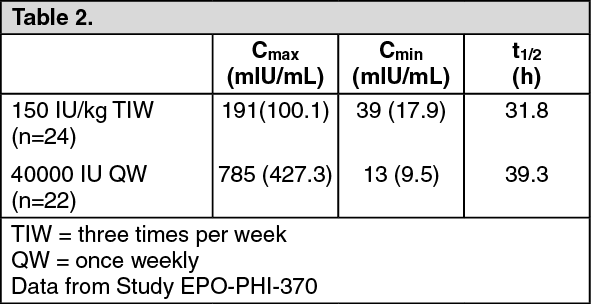 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image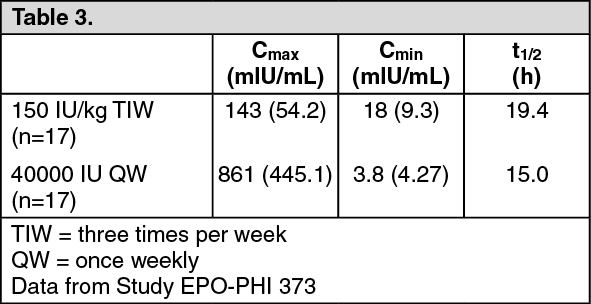 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image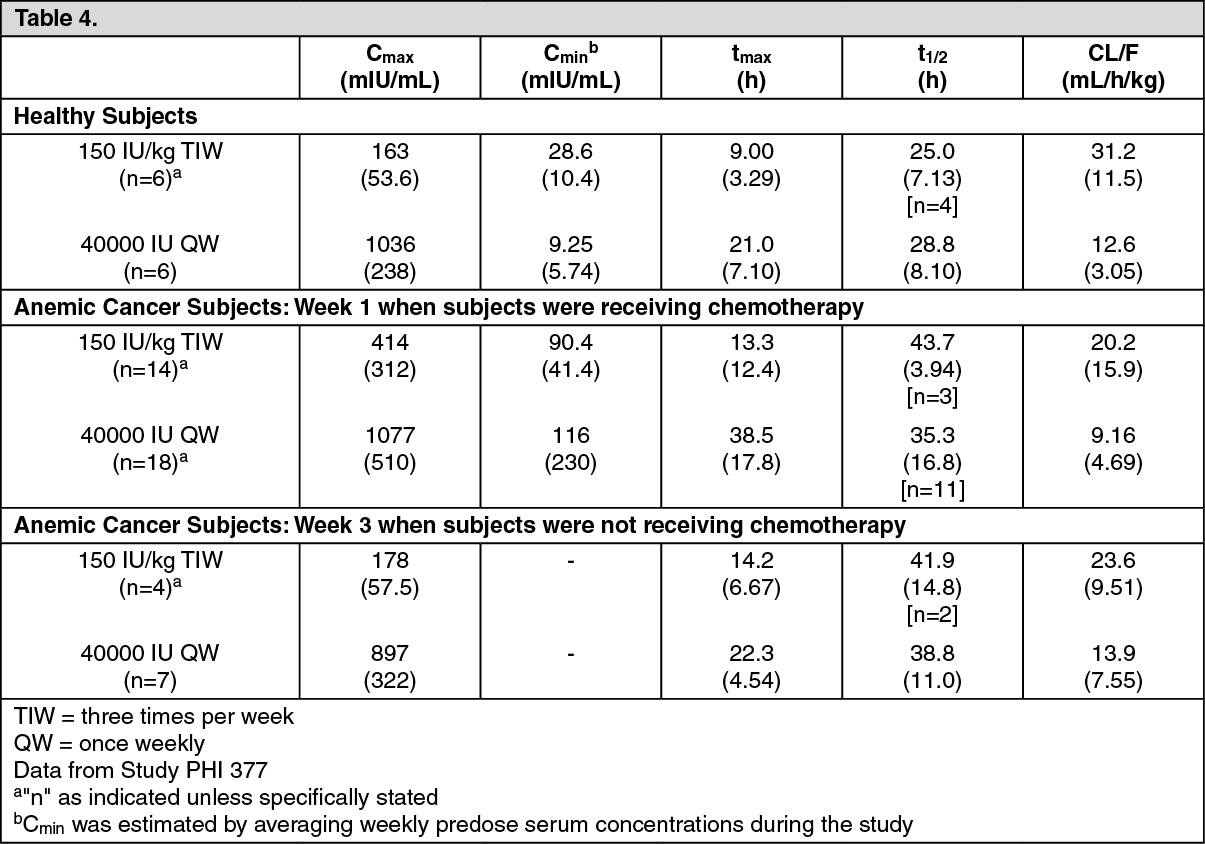 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image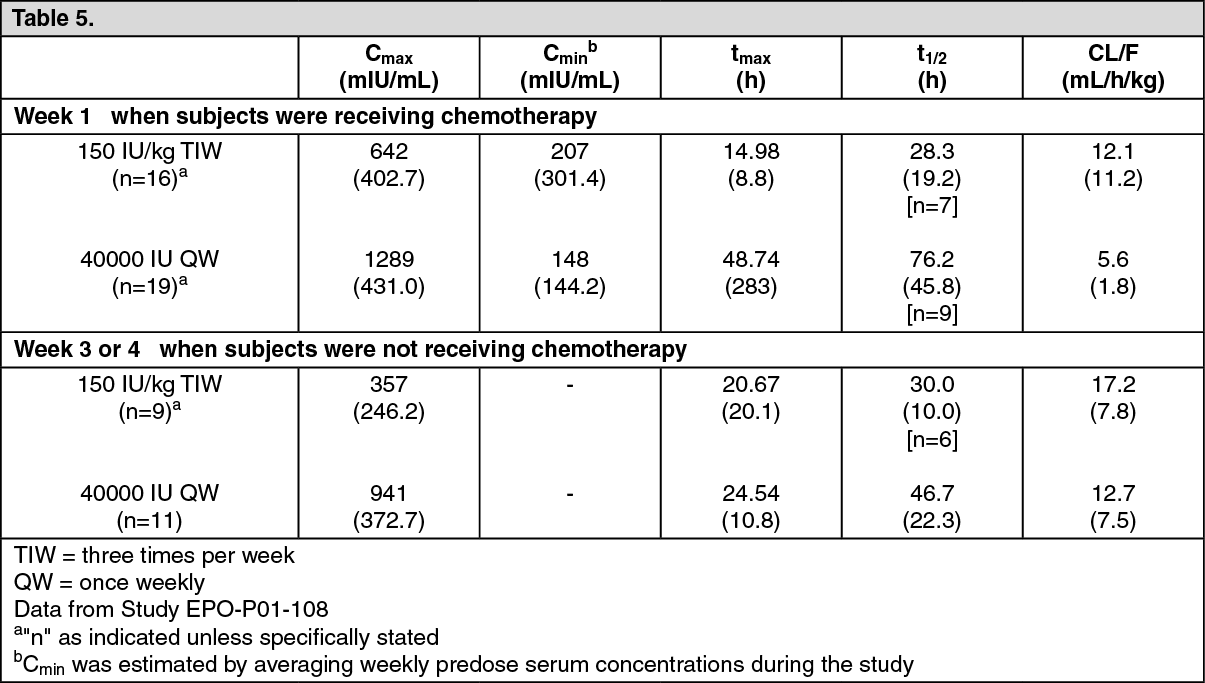 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image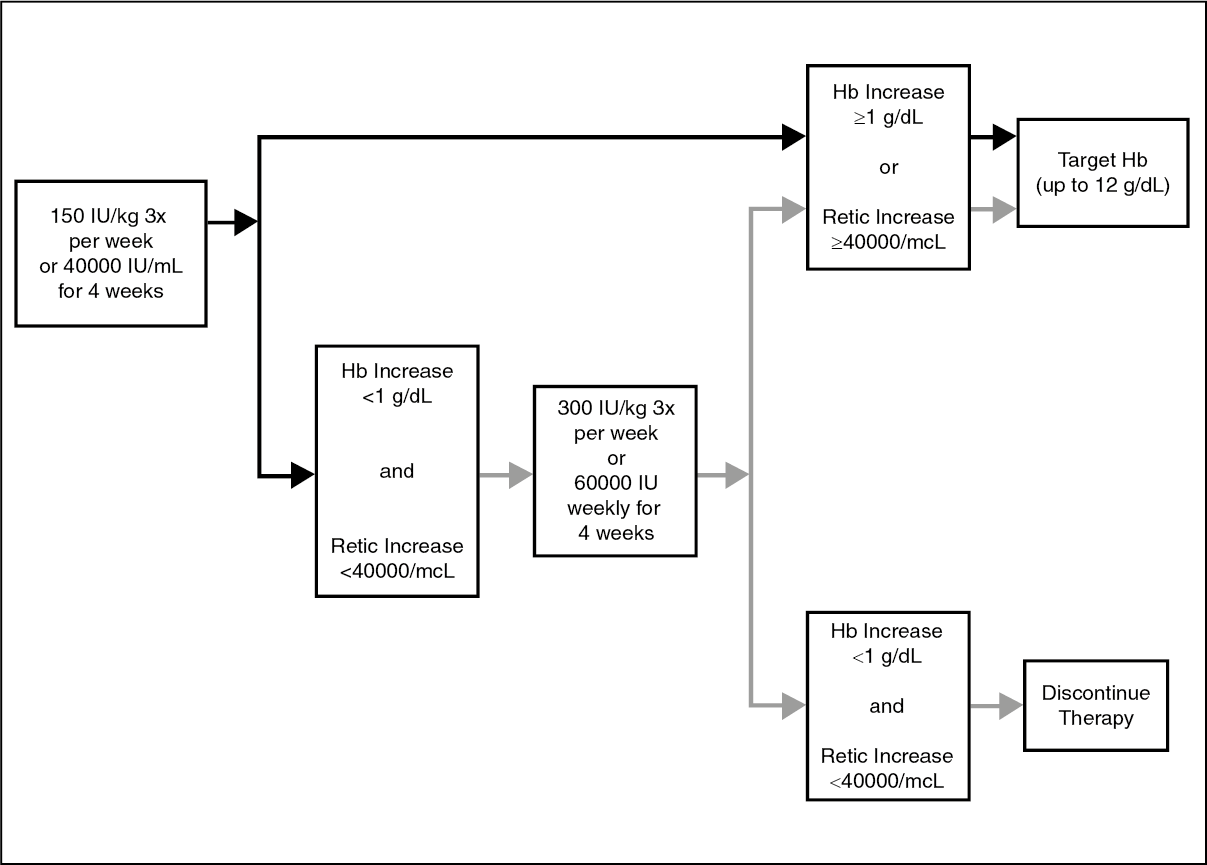 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image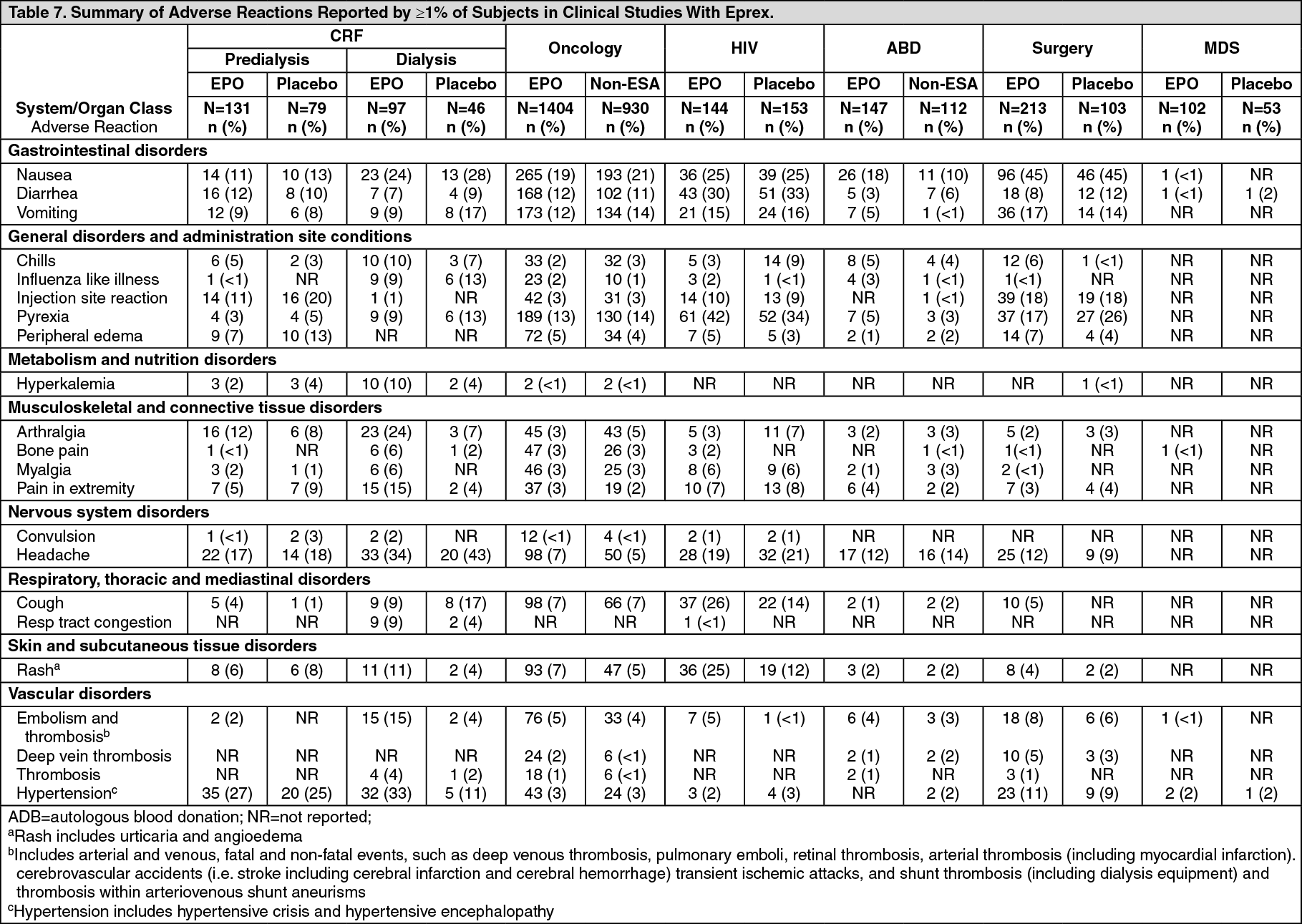 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 10_000 IU_mL147dc770-4790-4f6b-9725-a7ac00ad4d91.GIF)
 2_000 IU_0.5 mL9805b268-5e56-4482-8b40-a7ac00ad4d9f.GIF)
 4_000 IU_0.4 mL276ffcb2-ef2f-47d8-859e-a7ac00ad4dc9.GIF)
 40_000 IU_mLdf75797f-c623-4d02-b352-a92900ef9652.GIF)

 Sign Out
Sign Out
