Dosage: Treatment should be initiated under the supervision of a physician experienced in the treatment of hemophilia B.
Treatment with all factor IX products, including BeneFIX, requires individualized dosage adjustment. The dosage and duration of treatment for all factor IX products depends on the severity of the factor IX deficiency, the location and extent of bleeding, and the patient's clinical condition. Dosing of BeneFIX may differ from that of plasma-derived factor IX products.
To ensure that the desired factor IX activity level has been achieved, precise monitoring using the factor IX activity assay is advised, in particular for surgical interventions. In order to adjust the dose as appropriate, doses should be titrated taking into consideration factor IX activity, pharmacokinetic parameters (such as half-life and recovery) as well as the clinical situation.
The number of units of factor IX administered is expressed in International Units (IU), which are related to the current WHO standard for factor IX products. Factor IX activity in plasma is expressed either as a percentage (relative to normal human plasma) or in IU (relative to an international standard for factor IX in plasma). One IU of factor IX activity is equivalent to that quantity of factor IX in one ml of normal human plasma. Pharmacokinetics have to be assessed regularly in each patient and dosing has to be adjusted accordingly.
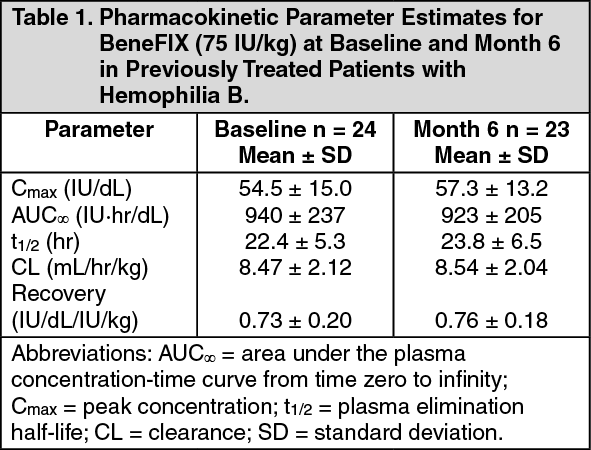
Patients ≥ 15 years: In patients ≥ 15 years, on average, one IU of BeneFIX per kilogram of body weight increased the circulating activity of factor IX by 0.8 ± 0.2 (range 0.4 to 1.4) IU/dL. The method of dose estimation is illustrated in the following example. If you use 0.8 IU/dL average increase of factor IX per IU/kg body weight administered, then: (see Equation 1).
 Click on icon to see table/diagram/image
Pediatric Patients (<15 years):
Click on icon to see table/diagram/image
Pediatric Patients (<15 years): In pediatric patients, on average, one international unit of BeneFIX per kilogram of body weight increased the circulating activity of factor IX by 0.7 ± 0.3 (range 0.2 to 2.1) IU/dL. The method of dose estimation is illustrated in the following example. If the patient use 0.7 IU/dL average increase of factor IX per IU/kg body weight administered, then: (see Equation 2).
Click on icon to see table/diagram/image
Dosing for Bleeding Episodes and Surgery: In the case of hemorrhagic events, the factor IX activity should not fall below the given plasma activity levels (in % of normal or in IU/dL) in the corresponding period. (See Table 3.)
Click on icon to see table/diagram/image
Dosage for Prophylaxis: For long term prophylaxis against bleeding in patients with severe haemophilia B, BeneFIX may be administered. In a clinical study for routine secondary prophylaxis the average dose for previously treated adult patients (PTP) was 40 IU/kg (range 13 to 78 IU/kg) at intervals of 3 to 4 days. In younger patients, shorter dosage intervals or higher doses may be necessary.
Use in Pediatric: Data from BeneFIX safety, efficacy, and pharmacokinetic studies have been evaluated in previously treated and previously untreated pediatric patients.
Nineteen (19) previously treated pediatric patients (range 4 to <15 years) underwent pharmacokinetic evaluations for up to 24 months. The mean increase in circulating factor IX activity was 0.7 ± 0.2 IU/dL per IU/kg infused (range 0.3 to 1.1 IU/dL per IU/kg). The mean biological half-life was 20.2 ± 4.0 hours (range 14 to 28 hours).
Fifty-eight previously untreated patients [PUPs] less than 15 years of age at baseline [3 neonates (0- <1 month), 45 infants (≥1 month-<2 years), 9 children (≥2 years-<12 years) and 1 adolescent >12 years)] underwent at least one recovery assessment within 30 minutes post-infusion in the presence or absence of hemorrhage during the study. The mean increase in circulating FIX activity was 0.7 ± 0.3 IU/dL per IU/kg infused (range 0.2 to 2.1 IU/dL per IU/kg). In addition, there was no difference in the recoveries noted when data were evaluated by age group for infants (0.7 ± 0.4 IU/dL per IU/kg; range 0.2 to 2.1 IU/dL per IU/kg) and children (0.7 ± 0.2 IU/dL per IU/kg; range 0.2 to 1.5 IU/dL per IU/kg). The recoveries in these age groups were consistent with the recovery for the PUP study as a whole. There was insufficient sample size in the neonate and adolescent age groups to perform an analysis in these groups. Data from 57 patients who underwent repeat recovery testing for up to 60 months demonstrated that the average incremental FIX recovery was consistent over time.
Elderly population: Clinical studies of BeneFIX did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. As with any patient receiving BeneFIX, dose selection for an elderly patient should be individualized.
Method of administration: BeneFIX is administered intravenously after reconstitution of the lyophilised powder for solution for injection with the supplied diluent (see section 6.6). It should be injected over several minutes. The rate of administration should be determined by the patient's comfort level.
Attach the syringe to the luer end of the infusion set tubing provided.
Apply a tourniquet and prepare the injection site by wiping the skin well with an alcohol swab provided in the kit.
Perform venipuncture. Insert the needle on the infusion set tubing into the vein, and remove the tourniquet. The reconstituted BeneFIX product should be injected intravenously over several minutes. The rate of administration should be determined by the patient's comfort level.
Reconstituted BeneFIX should not be administered in the same tubing or container with other medicinal products.
Following completion of BeneFIX treatment, remove the infusion set and discard (see Special precautions for disposal and other handling under Cautions for Usage).
The safety and efficacy of administration by continuous infusion have not been established. See also Precautions and Adverse Reactions.
BeneFIX should be administered using the infusion set provided in the kit, and the pre-filled diluent syringe provided or a single, sterile, disposable plastic syringe. In addition, the solution should be withdrawn from the vial using the vial adapter.
The reconstituted solution may be stored at room temperature prior to administration. However, BeneFIX should be administered within 3 hours after reconstitution.
Note: Agglutination of red blood cells in the tubing/syringe has been reported with the administration of BeneFIX. No adverse events have been reported in association with this observation. To minimize the possibility of agglutination, it is important to limit the amount of blood entering the tubing. Blood should not enter the syringe. If red blood cell agglutination is observed in the tubing or syringe, discard all material (tubing, syringe and BeneFIX solution) and resume administration with a new package.
If any suspected hypersensitivity reaction takes place that is thought to be related to the administration of BeneFIX, the rate of infusion should be decreased or the infusion stopped (see Precautions and Adverse Reactions).
Reconstitution: Always wash hands before performing the following procedures. Aseptic technique (meaning clean and germ-free) should be used during the reconstitution procedure. All components used in the reconstitution and administration of this product should be used as soon as possible after opening their sterile containers to minimize unnecessary exposure to the atmosphere.
BeneFIX is administered by IV infusion after reconstitution with the supplied diluent (0.234% sodium chloride diluent) in the pre-filled syringe.
Allow the vial of lyophilized BeneFIX and the pre-filled diluent syringe to reach room temperature.
Remove the plastic flip-top cap from the BeneFIX vial to expose the central portions of the rubber stopper.
Wipe the top of the vial with the alcohol swab provided, or use another antiseptic solution, and allow to dry. After cleaning, do not touch the rubber stopper with your hand or allow it to touch any surface.
Peel back the cover from the clear plastic vial adapter package.
Do not remove the adapter from the package. Place the vial on a flat surface. While holding the adapter in the package, place the vial adapter over the vial. Press down firmly on the package until the adapter snaps into place on the top of the vial, with the adapter spike penetrating the vial stopper. Leave the adapter package in place.
Grasp the plunger rod as shown in the diagram. Avoid contact with the shaft of the plunger rod. Attach the threaded end of the plunger rod to the diluent syringe plunger by pushing and turning firmly. Remove the tamper-resistant, plastic-tip cap from the diluent syringe by bending the cap up and down to break the perforation. Do not touch the inside of the cap or the syringe tip. Place the cap on its side on a clean surface in a spot where it would be least likely to become environmentally contaminated.
Lift the package away from the adapter and discard the package.
Place the vial on a flat surface. Connect the diluent syringe to the vial adapter by inserting the tip into the adapter opening while firmly pushing and turning the syringe clockwise until the connection is secured.
Slowly depress the plunger rod to inject all the diluent into the BeneFIX vial.
Without removing the syringe, gently swirl the contents of the vial until the powder is dissolved.
Inspect the final solution for specks before administration. The solution should appear clear and colorless.
Note: If you use more than one vial of BeneFIX per infusion, reconstitute each vial by following the previous instructions.
Ensuring that the syringe plunger rod is still fully depressed, invert the vial. Slowly draw the solution into the syringe.
Note: If you prepared more than one vial of BeneFIX, remove the diluent syringe from the vial adapter, leaving the vial adapter attached to the vial. Quickly attach a separate large luer lock syringe and draw back the reconstituted contents as instructed above. Repeat this procedure with each vial in turn. Do not detach the diluent syringes or the large luer lock syringe until you are ready to attach the large luer lock syringe to the next vial adapter.
Detach the syringe from the vial adapter by gently pulling and turning the syringe counterclockwise. Discard the vial with the adapter attached.
Note: If the solution is not to be used immediately, the syringe cap should be carefully replaced. Do not touch the syringe tip or the inside of the cap.
BeneFIX should be administered within 3 hours after reconstitution. The reconstituted solution may be stored at room temperature prior to administration.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image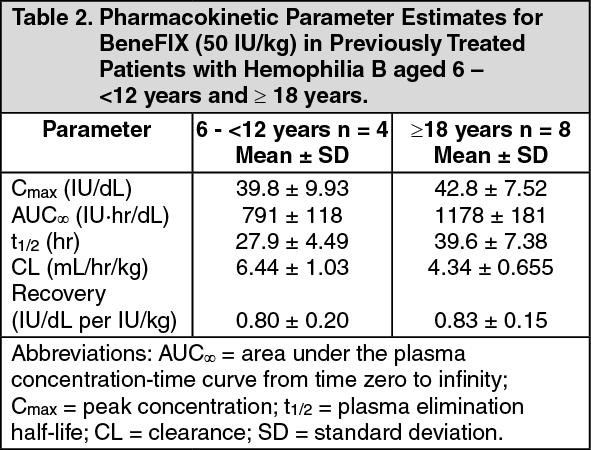 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image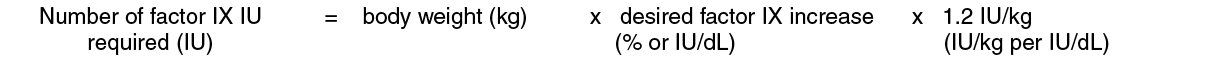 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image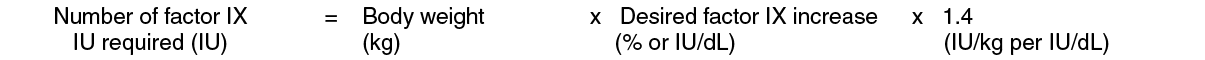 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image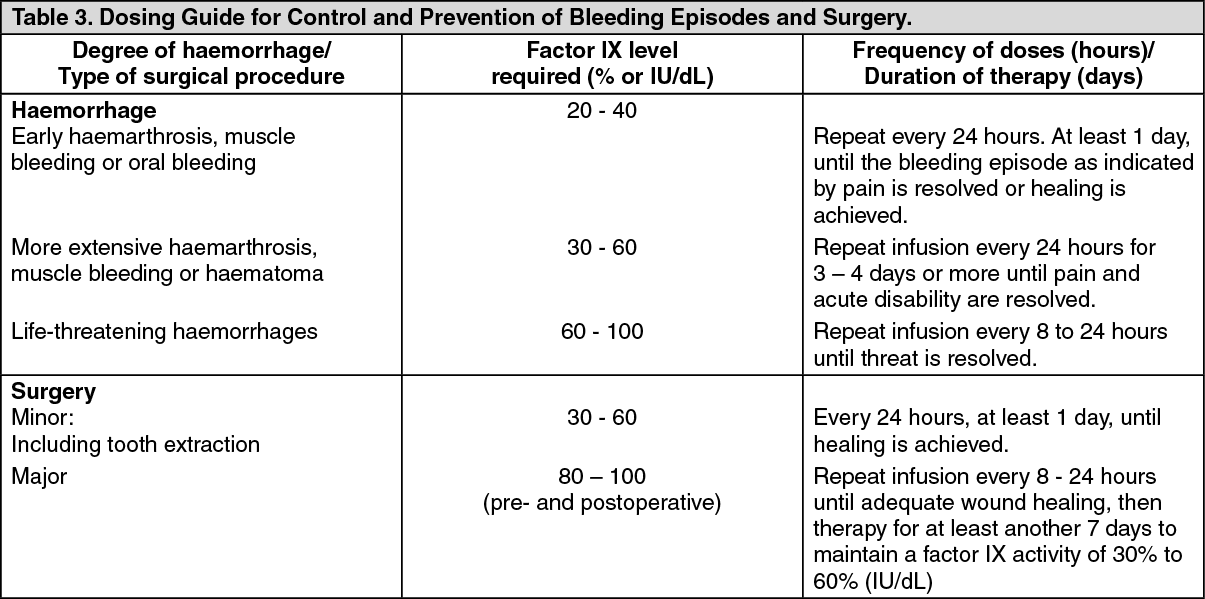 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image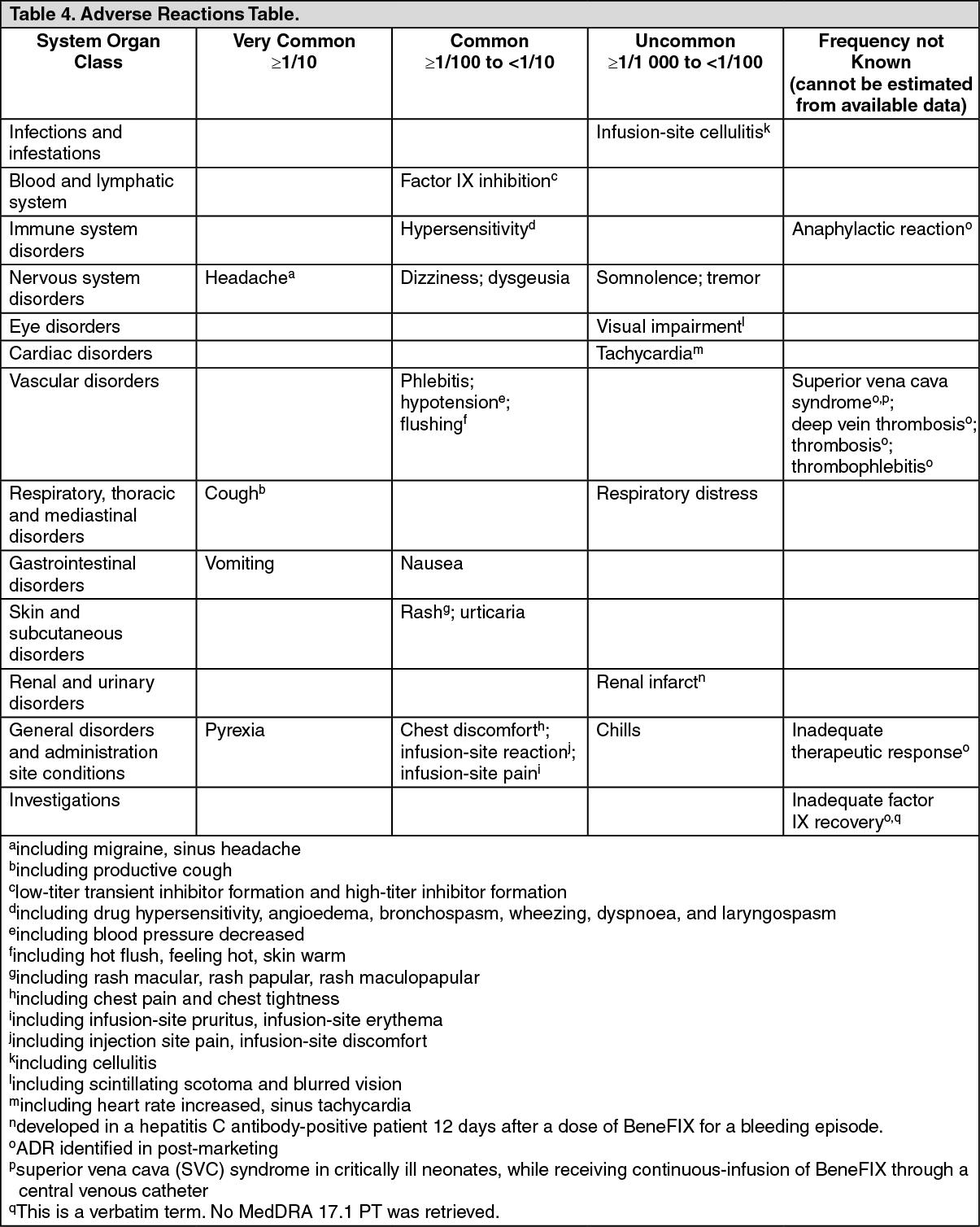 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
