Đăng xuất
Đăng xuất

Acalabrutinib là một chất ức chế Bruton's tyrosine kinase (BTK) thuộc nhóm phân tử nhỏ. Acalabrutinib và chất chuyển hóa có hoạt tính là ACP-5862, tạo thành một liên kết cộng hóa trị với nhánh cystein trong vị trí hoạt động của BTK, dẫn đến ức chế hoạt tính enzyme của BTK. BTK là một phân tử truyền tín hiệu của thụ thể kháng nguyên tế bào B (BCR) và con đường thụ thể cytokin. Ở tế bào B, tín hiệu BTK làm kích hoạt con đường cần thiết cho sự tăng sinh, di chuyển, hóa ứng động và thâm nhập mô của tế bào B. Trong nghiên cứu tiền lâm sàng, acalabrutinib ức chế hoạt hóa các protein tín hiệu điều hòa xuôi dòng CD86 và CD69 qua trung gian BTK và ức chế tăng sinh tế bào B ác tính và ức chế sự tăng trưởng của khối u trong mô hình cấy ghép ung thư trên chuột.
Đặc tính dược lực học
Nhóm dược lý trị liệu: tác nhân chống ung thư, các chất ức chế protein kinase, mã ATC: L01EL02.
Ở những bệnh nhân có tế bào B ác tính dùng liều 100 mg mỗi 12 giờ, trạng thái ổn định trung vị của BTK chiếm ≥ 95% trong máu ngoại vi được duy trì hơn 12 giờ, gây bất hoạt BTK trong khoảng thời gian dùng thuốc theo khuyến cáo.
Điện sinh lý học tim
Hiệu quả của acalabrutinib trên khoảng QTc đã được đánh giá trong một thử nghiệm ngẫu nhiên, mù đôi, giả đôi, đối chứng với thuốc và giả dược, bắt chéo 4 chiều về khoảng QTc ở 48 đối tượng là người trưởng thành khỏe mạnh. Việc sử dụng một liều acalabrutinib duy nhất cao gấp 4 lần liều tối đa khuyến cáo không gây ra kéo dài khoảng QTc ở bất kỳ mức độ ý nghĩa lâm sàng nào (ví dụ ≥ 10 miligiây).
Đặc tính dược động học
Acalabrutinib có dược động học tỷ lệ với liều, cả acalabrutinib và chất chuyển hóa có hoạt tính ACP-5862 đều tăng tiếp xúc trong khoảng liều từ 75 đến 250 mg (tương đương 0,75 đến 2,5 lần liều đơn khuyến cáo) ở bệnh nhân có bệnh lý tế bào B ác tính. Với liều khuyến cáo 100 mg hai lần/ngày, trung bình nhân (% hệ số biến thiên - CV) của diện tích dưới đường cong của nồng độ thuốc trong huyết tương theo thời gian (AUC24h) và nồng độ tối đa trong huyết tương (Cmax) của acalabrutinib lần lượt là 1.843 ng•h/mL (38%) và 563 ng/mL (29%), và đối với ACP-5862 lần lượt là 3.947 ng•h/mL (43%) và 451 ng/mL (52%).
Hấp thu
Trung bình nhân sinh khả dụng tuyệt đối của acalabrutinib là 25%. Trung vị [giá trị nhỏ nhất, giá trị lớn nhất] của thời gian đạt được nồng độ đỉnh của acalabrutinib trong huyết tương (Tmax) là 0,9 giờ [0,5; 1,9] và ACP-5862 là 6 giờ [0,9; 2,7].
Tác động của thức ăn
Ở người khỏe mạnh, sử dụng một liều đơn 75 mg acalabrutinib (0,75 lần so với liều đơn khuyến cáo) cùng với bữa ăn giàu chất béo, nhiều calorie (khoảng 918 calorie, 59 g carbohydrat, 59 g chất béo và 39 g protein) không có ảnh hưởng đến AUC trung bình so với dùng thuốc khi đói. Tuy nhiên, Cmax giảm 73% và Tmax bị trì hoãn 1-2 giờ.
Phân bố
Acalabrutinib và ACP-5862 gắn thuận nghịch với protein huyết tương người với tỷ lệ lần lượt là 97,5% và 98,6%. Tỷ lệ trung bình trong máu/huyết tương in vitro là 0,8 đối với acalabrutinib và 0,7 đối với ACP-5862. Trung bình nhân (% CV) của thể tích phân bố ở trạng thái ổn định (Vss) là khoảng 101 L (52%) đối với acalabrutinib và 67 L (32%) đối với ACP-5862.
Thanh thải
Trung bình nhân (% CV) của thời gian bán thải (t1/2) là 1 giờ (59%) đối với acalabrutinib và 3,5 giờ (24%) đối với ACP-5862. Trung bình nhân (% CV) của độ thanh thải biểu kiến đường uống (CL/F) là 71 L/giờ (35%) đối với acalabrutinib và 13 L/giờ (42%) đối với ACP-5862.
Chuyển hóa
Dựa trên kết quả các nghiên cứu in vitro, acalabrutinib được chuyển hóa chủ yếu bởi các enzym CYP3A và một phần rất nhỏ thông qua quá trình liên hợp glutathion và thủy phân amid. ACP-5862 được xác định là chất chuyển hóa có hoạt tính chính trong huyết tương với mức phơi nhiễm trung bình (AUC) cao hơn khoảng 2 đến 3 lần so với acalabrutinib. ACP-5862 có hoạt tính ức chế BTK thấp hơn khoảng 50% so với acalabrutinib.
Thải trừ
Sau khi dùng một liều 100 mg duy nhất acalabrutinib có đánh dấu đồng vị phóng xạ ở người khỏe mạnh, 84% liều dùng đã được tìm thấy trong phân và 12% liều được thải trừ qua nước tiểu, và ít hơn 2% lượng liều được bài tiết dưới dạng acalabrutinib ở dạng nguyên vẹn không chuyển hóa trong nước tiểu và phân.
Nhóm bệnh nhân đặc biệt
Tuổi, chủng tộc và cân nặng
Tuổi (32 đến 90 tuổi), giới tính, chủng tộc (người da trắng, người Mỹ gốc Phi) và cân nặng (40 đến 149 kg) không có ảnh hưởng có ý nghĩa lâm sàng đến đặc tính dược động học của acalabrutinib và chất chuyển hóa còn hoạt tính ACP-5862.
Bệnh nhân suy thận
Không thấy sự khác biệt về đặc tính dược động học liên quan đến lâm sàng ở bệnh nhân suy thận nhẹ hoặc trung bình (eGFR ≥ 30 mL/phút/1,73m2, ước tính bằng công thức MDRD (công thức hiệu chỉnh chế độ ăn đối với bệnh thận). Đặc tính dược động học của acalabrutinib chưa được đánh giá ở bệnh nhân có suy thận nặng (eGFR < 29 mL/phút/ 1,73m2, ước tính bằng MDRD) hoặc bệnh nhân suy thận cần lọc máu.
Bệnh nhân suy gan
AUC của acalabrutinib tăng gấp 1,9 lần ở những người suy gan nhẹ (Child-Pugh A), tăng 1,5 lần ở những người suy gan trung bình (Child-Pugh B) và gấp 5,3 lần ở những người có suy gan nặng (Child-Pugh C) so với người có chức năng gan bình thường. Quan sát không thấy sự khác biệt liên quan đến lâm sàng về đặc tính dược động học của ACP-5862 ở những người suy gan nặng (Child-Pugh C) so với người có chức năng gan bình thường. Không thấy sự khác biệt liên quan đến lâm sàng về đặc tính dược động học của acalabrutinib và ACP-5862 ở những bệnh nhân suy gan nhẹ hoặc trung bình (bilirubin toàn phần ít hơn và bằng giới hạn bình thường trên [ULN] và AST cao hơn ULN, hoặc bilirubin toàn phần cao hơn ULN với mọi giá trị của AST) so với nhóm bệnh nhân có chức năng gan bình thường (bilirubin toàn phần và AST nằm trong ULN).
Các nghiên cứu về tương tác thuốc
Tác động của các thuốc ức chế CYP3A đến acalabrutinib
Sử dụng đồng thời với các thuốc ức chế mạnh CYP3A (ví dụ itraconazol 200 mg 1 lần/ngày trong 5 ngày) làm tăng giá trị Cmax của acalabrutinib gấp 3,9 lần và tăng AUC lên 5,1 lần ở người khỏe mạnh.
Mô hình mô phỏng dược động học dựa trên sinh lý (PBPK) của acalabrutinib với các thuốc ức chế CYP3A mức độ trung bình (erythromycin, fluconazole và diltiazem) đã cho thấy việc sử dụng đồng thời làm tăng giá trị Cmax và AUC của acalabrutinib khoảng từ 2 đến 3 lần.
Tác động của các thuốc cảm ứng CYP3A đến acalabrutinib
Sử dụng đồng thời với các thuốc cảm ứng mạnh CYP3A (như rifampin 600 mg 1 lần/ngày trong 9 ngày) làm giảm giá trị Cmax của acalabrutinib 68% và AUC 77% ở người khỏe mạnh.
Các thuốc làm giảm acid dịch vị
Độ tan của acalabrutinib giảm khi pH tăng. Việc sử dụng đồng thời với một antacid (ví dụ 1 g calci carbonat) làm giảm giá trị AUC của acalabrutinib đi 53% ở người khỏe mạnh. Việc sử dụng đồng thời với một chất ức chế bơm proton (như omeprazol liều 40 mg trong 5 ngày) làm giảm giá trị AUC của acalabrutinib mất 43%.
Các nghiên cứu in vitro
Các con đường chuyển hóa
Acalabrutinib là một chất ức chế yếu CYP3A4/5, CYP2C8 và CYP2C9, nhưng không có tác dụng ức chế CYP1A2, CYP2B6, CYP2C19, CYP2D6, UGT1A1, và UGT2B7. ACP-5862 là một chất ức chế yếu CYP2C8, CYP2C9 và CYP2C19, nhưng không ức chế CYP1A2, CYP2B6, CYP2D6, CYP3A4/5, UGT1A1 và UGT2B7.
Acalabrutinib là một chất cảm ứng yếu CYP1A2, CYP2B6 và CYP3A4; ACP-5862 là chất cảm ứng yếu CYP3A4.
Dựa trên dữ liệu in vitro và mô hình PBPK, không có tương tác với chất nền của CYP ở nồng độ phù hợp lâm sàng.
Các hệ thống vận chuyển thuốc
Acalabrutinib và chất chuyển hóa có hoạt tính ACP-5862 là cơ chất của glycoprotein P (P-gp) và của protein kháng ung thư vú (BCRP). Acalabrutinib không phải là cơ chất của các chất vận chuyển tái hấp thu ở thận như OAT1, OAT3 và OCT2, hoặc các chất vận chuyển tại gan như OATP1B1 và OATP1B3. ACP-5862 không phải là chất nền của OATP1B1 hoặc OATP1B3.
Acalabrutinib và ACP-5862 không ức chế P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 và MATE2-K ở nồng độ phù hợp lâm sàng.
Acalabrutinib có thể làm tăng tiếp xúc với các chất nền đồng vận chuyển qua BCRP (ví dụ, methotrexat) bằng cách ức chế BCRP ở ruột. ACP-5862 không ức chế BCRP ở nồng độ phù hợp lâm sàng. Acalabrutinib không ức chế MATE1, trong khi ACP-5862 có thể làm tăng tiếp xúc với chất nền đồng vận chuyển của MATE1 (ví dụ, metformin) bằng cách ức chế MATE1.
NGHIÊN CỨU LÂM SÀNG
U lympho tế bào vỏ
Hiệu quả của CALQUENCE được dựa theo kết quả của Thử nghiệm LY-004 với tên gọi “Nghiên cứu pha 2, nhãn mở về ACP-196 trên đối tượng có bệnh u lympho tế bào vỏ” (NCT02213926). Thử nghiệm LY-004 được tiến hành trên tổng cộng 124 bệnh nhân MCL đã được nhận ít nhất một liệu pháp điều trị trước đây.
Tuổi trung vị của bệnh nhân nghiên cứu là 68 tuổi (dao động từ 42 đến 90), 80% là nam và 74% là người da trắng. 93% bệnh nhân có điểm số thể trạng ban đầu ECOG là 0 hoặc 1. Trung vị thời gian tính từ khi có chẩn đoán là 46,3 tháng và số trung vị lần điều trị trước đây là 2 lần (từ khoảng 1 đến 5 lần), bao gồm 18% đã có ghép tế bào gốc trước đó. Những bệnh nhân đã từng được điều trị trước đây bằng thuốc ức chế BTK không được chọn vào nghiên cứu. Các phác đồ được dùng phổ biến nhất là phác đồ CHOP (52%) và ARA-C (34%). Trước khi dùng thuốc nghiên cứu, 37% bệnh nhân có ít nhất một khối u với đường kính dài nhất ≥ 5 cm, 73% đã có ung thư lan sang các hạch, trong đó 51% có liên quan đến tủy xương. Điểm MIPI giản lược (bao gồm tuổi, điểm ECOG, lactat dehydrogenase nền và số lượng bạch cầu) ở mức trung bình với 44% và mức cao với 17% bệnh nhân.
CALQUENCE được dùng đường uống với liều 100 mg mỗi 12 giờ cho đến khi bệnh tiến triển hoặc xuất hiện độc tính không thể dung nạp được. Mức cường độ liều trung vị là 98,5%. Kết quả chính về hiệu quả trong Thử nghiệm LY004 là tỷ lệ đáp ứng tổng thể và trung vị thời gian theo dõi là 15,2 tháng.
- xem Bảng 1.

Trung vị thời gian đạt được đáp ứng tốt nhất là 1,9 tháng.
Tăng tế bào lympho
Khi bắt đầu sử dụng CALQUENCE, thử nghiệm LY-004 ghi nhận 31,5% bệnh nhân có tăng số lượng tế bào lympho thoáng qua (được định nghĩa là số lượng tế bào lympho tuyệt đối (ALC) tăng ≥ 50% so với trước điều trị và sau điều trị cho giá trị ≥ 5x109). Trung vị thời gian bắt đầu có tăng lympho bào là 1,1 tuần và trung vị khoảng thời gian của tăng lympho bào là 6,7 tuần.
Bệnh bạch cầu mạn dòng lympho
Hiệu lực của CALQUENCE ở bệnh nhân CLL đã được chứng minh trong hai thử nghiệm ngẫu nhiên, có đối chứng. CALQUENCE cũng được chỉ định cho SLL vì đây là bệnh tương tự như CLL.
Thử nghiệm ELEVATE-TN
Hiệu lực của CALQUENCE được đánh giá trong thử nghiệm ELEVATE-TN, một thử nghiệm ngẫu nhiên, đa trung tâm, nhãn mở, có đối chứng với thuốc (có hoạt chất), với 3 nhánh nghiên cứu sử dụng CALQUENCE phối hợp với obinutuzumab, CALQUENCE đơn trị liệu và obinutuzumab kết hợp với chlorambucil ở 535 bệnh nhân mắc bệnh bạch cầu mạn dòng lympho chưa từng điều trị trước đây (NCT02475681). Bệnh nhân ≥ 65 tuổi hoặc từ 18 đến 65 tuổi với tổng điểm theo thang đánh giá bệnh tích lũy (CIRS) > 6 hoặc độ thanh thải creatinin từ 30 đến 69 mL/phút được đưa vào nghiên cứu. Thử nghiệm cũng yêu cầu mức transaminase gan ≤ 3 lần giới hạn bình thường trên (ULN) và bilirubin toàn phần ≤ 1,5 lần ULN và loại trừ bệnh nhân có chuyển đổi Richter.
Bệnh nhân được chia ngẫu nhiên theo tỷ lệ 1: 1: 1 vào 3 nhánh sử dụng:
• CALQUENCE phối hợp với obinutuzumab (CALQUENCE+G): dùng CALQUENCE 100 mg mỗi 12 giờ bắt đầu từ Chu kỳ 1 ngày 1 cho đến khi bệnh tiến triển hoặc xuất hiện độc tính không thể dung nạp được. Obinutuzumab được dùng bắt đầu từ Chu kỳ 2 ngày 1 đến tối đa 6 chu kỳ điều trị. Dùng obinutuzumab liều 1.000 mg vào các ngày 1 và 2 (liều 100 mg ngày 1 và 900 mg ngày 2), ngày 8 và 15 Chu kỳ 2, tiếp đến là 1.000 mg ngày 1 của chu kỳ 3 cho đến 7. Mỗi chu kỳ kéo dài 28 ngày.
• CALQUENCE đơn trị liệu: dùng CALQUENCE liều 100 mg mỗi 12 giờ cho đến khi bệnh tiến triển hoặc xuất hiện độc tính không thể dung nạp được.
• Obinutuzumab phối hợp với chlorambucil (GClb): điều trị với obinutuzumab và chlorambucil tối đa 6 chu kỳ điều trị. Truyền tĩnh mạch obinutuzumab liều 1.000 mg vào Ngày 1 và 2 (liều 100 mg vào Ngày 1 và 900 mg vào Ngày 2), ngày 8 và 15 chu kỳ 1 và được nối tiếp bằng 1.000 mg ngày 1 của Chu kỳ 2 đến 6. Chlorambucil liều 0,5 mg/kg được dùng đường uống vào Ngày 1 và 15 của Chu kỳ 1 đến 6. Mỗi chu kỳ kéo dài 28 ngày.
Bệnh nhân được phân ngẫu nhiên theo tình trạng đột biến mất đoạn 17p, chỉ số thể trạng theo ECOG (0 hoặc 1 so với 2) và khu vực địa lý. Tổng cộng có 535 bệnh nhân được chọn ngẫu nhiên, 179 vào nhóm CALQUENCE+G, 179 vào nhóm CALQUENCE đơn trị liệu và 177 vào nhóm GClb. Tuổi trung vị là 70 tuổi (từ 41 đến 91 tuổi), 47% có giai đoạn bệnh Rai là III hoặc IV, 14% có đột biến mất đoạn 17p hoặc đột biến TP53, 63% bệnh nhân có IGVH không đột biến và 18% mất đoạn 11q. Đặc điểm nhân khẩu học và bệnh lý nền là tương tự nhau giữa các nhánh điều trị.
Hiệu lực dựa trên thời gian sống không bệnh tiến triển (PFS) được đánh giá bởi Hội đồng thẩm định độc lập (IRC). Thời gian trung vị theo dõi là 28,3 tháng (dao động từ 0,0 đến 40,8 tháng). Kết quả về hiệu lực được trình bày trong Bảng 2. Đường cong Kaplan-Meier đối với PFS được biểu diễn trong Hình 1.
- xem Bảng 2 & Hình 1.

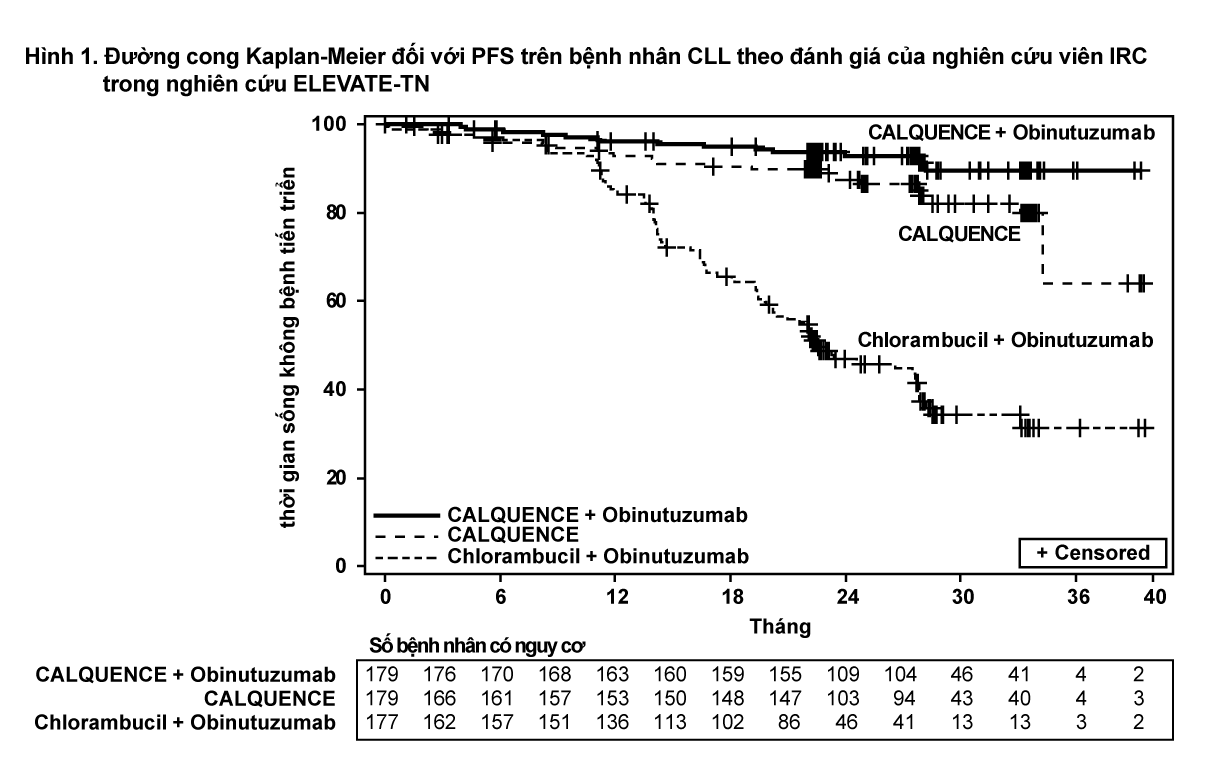
Với thời gian trung vị theo dõi là 28,3 tháng, trung vị thời gian sống còn toàn bộ không đạt được ở mỗi nhánh, với ít hơn 10% số bệnh nhân được ghi nhận có biến cố.
Thử nghiệm ASCEND
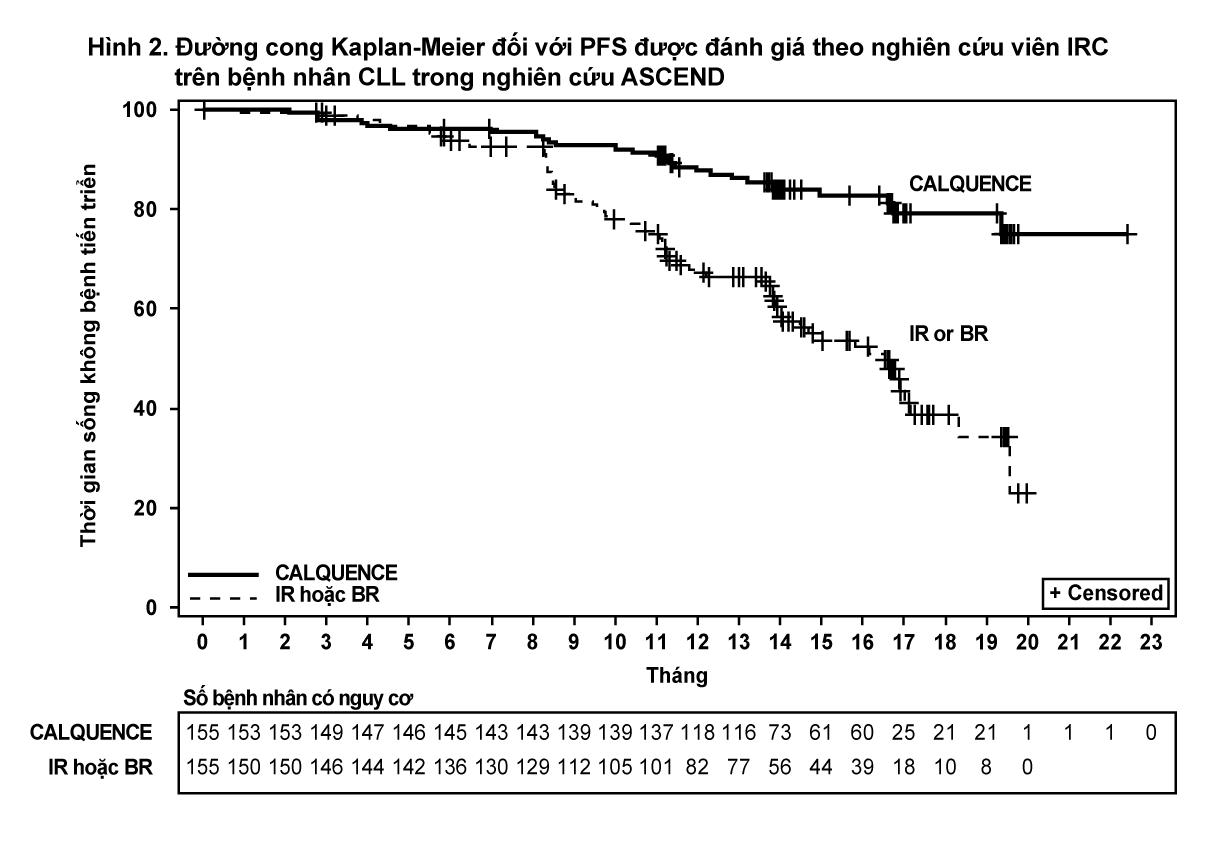
Hiệu lực của CALQUENCE ở bệnh nhân CLL tái phát hoặc kháng trị được dựa trên kết quả một thử nghiệm nhãn mở, đa trung tâm, ngẫu nhiên (ASCEND; NCT02970318). Thử nghiệm đã thu nhận 310 bệnh nhân CLL tái phát hoặc kháng trị đã từng điều trị ít nhất một liệu pháp toàn thân trước đây. Thử nghiệm không bao gồm những bệnh nhân có chuyển đổi thể bệnh, bệnh bạch cầu tiền lympho bào, hoặc đã điều trị trước đó bằng venetoclax, một chất ức chế Bruton tyrosin kinase hoặc thuốc ức chế phosphoinositid-3 kinase.
Bệnh nhân được phân ngẫu nhiên theo tỷ lệ 1:1 vào nhóm:
• Dùng CALQUENCE 100 mg mỗi 12 giờ cho đến khi bệnh tiến triển hoặc xuất hiện độc tính không thể dung nạp được, hoặc
• Lựa chọn của nghiên cứu viên:
+ Idelalisib phối hợp với rituximab (IR): idelalisib uống 150 mg mỗi 12 giờ cho đến khi bệnh tiến triển hoặc xuất hiện độc tính không thể dung nạp được, kết hợp với truyền rituximab 8 lần (375 mg/m2 đường tĩnh mạch vào Ngày 1 của Chu kỳ 1, sau đó tiêm tĩnh mạch 500 mg/m2 sau mỗi 2 tuần cho 4 liều và sau đó cho 3 liều với mỗi 4 tuần), với độ dài một chu kỳ là 28 ngày.
+ Bendamustin phối hợp với rituximab (BR): bendamustin liều 70 mg/m2 (Ngày 1 và 2 của mỗi chu kỳ 28 ngày), kết hợp với truyền tĩnh mạch rituximab (375 mg/m2 vào Ngày 1 của Chu kỳ 1, sau đó 500 mg/m2 vào ngày 1 của các chu kỳ tiếp theo), đến tối đa 6 chu kỳ.
Bệnh nhân được phân ngẫu nhiên theo tình trạng đột biến mất đoạn 17p, chỉ số thể trạng theo ECOG (0 hoặc 1 so với 2) và số lượng phác đồ trước đó (1 đến 3 so với ≥ 4). Trong số 310 bệnh nhân, 155 được phân vào nhóm dùng CALQUENCE đơn trị liệu, 119 vào nhóm IR và 36 vào nhóm BR. Tuổi trung vị là 67 tuổi (từ 32 đến 90 tuổi), 42% có giai đoạn bệnh Rai là III hoặc IV, 28% có đột biến mất đoạn 17p hoặc đột biến TP53, 78% bệnh nhân có IGVH không đột biến và 27% có mất đoạn 11q. Nhóm sử dụng CALQUENCE có trung vị số liệu pháp được dùng trước đó là 1 (khoảng dao động là 1-8), với 47% có ít nhất 2 liệu pháp trước đó. Nhánh do nghiên cứu viên lựa chọn có trung vị số liệu pháp trước đó là 2 (dao động từ 1-10), với 57% bệnh nhân đã dùng ít nhất 2 liệu pháp trước đây.
Trong nhánh sử dụng CALQUENCE, trung vị thời gian điều trị là 15,7 tháng, với 94% bệnh nhân được điều trị ít nhất 6 tháng và 86% bệnh nhân được điều trị ít nhất 1 năm. Ở nhánh lựa chọn của nghiên cứu viên, trung vị thời gian điều trị là 8,4 tháng, với 59% số bệnh nhân được điều trị ít nhất 6 tháng và 37% được điều trị trong ít nhất 1 năm.
Hiệu lực dựa trên PFS được đánh giá bởi IRC, với trung vị thời gian theo dõi là 16,1 tháng (trong khoảng từ 0,03 đến 22,4 tháng). Kết quả về hiệu lực được trình bày trong Bảng 3. Đường cong Kaplan-Meier đối với PFS được biểu diễn ở Hình 2. Không có sự khác biệt có ý nghĩa thống kê về tỷ lệ đáp ứng tổng thể giữa hai nhánh điều trị.
- xem Bảng 3 & Hình 2.