Pharmacology: Pharmacodynamics: Mechanism of Action: Baloxavir marboxil is a prodrug that is converted by hydrolysis to its active metabolite, baloxavir, the active form that exerts anti-influenza activity. Baloxavir acts on the cap-dependent endonuclease (CEN), an influenza virus-specific enzyme in the polymerase acidic (PA) subunit of the viral RNA polymerase complex and thereby inhibits the transcription of influenza virus genomes resulting in inhibition of influenza virus replication. The 50% inhibition concentration (IC50) of baloxavir was 1.4 to 3.1 nmol/L for influenza A viruses and 4.5 to 8.9 nmol/L for influenza B viruses in an enzyme inhibition assay.
Nonclinical studies demonstrate potent antiviral activity of baloxavir against influenza A and B virus
in vitro and
in vivo. The antiviral activity of baloxavir against laboratory strains and clinical isolates of influenza A and B viruses was determined in the MDCK cell culture assay. The median 50% effective concentration (EC50) values of baloxavir were 0.73 nmol/L (n=31; range: 0.20-1.85 nmol/L) for subtype A/H1N1 strains, 0.83 nmol/L (n=33; range: 0.35-2.63 nmol/L) for subtype A/H3N2 strains, and 5.97 nmol/L (n=30; range: 2.67-14.23 nmol/L) for type B strains. In a MDCK cell-based virus titer reduction assay, the 90% effective concentration (EC90) values of baloxavir were in the range of 0.46 to 0.98 nmol/L for subtype A/H1N1 and A/H3N2 viruses, 0.80 to 3.16 nmol/L for avian subtype A/H5N1 and A/H7N9 viruses, and 2.21 to 6.48 nmol/L for type B viruses.
Viruses bearing the PA/I38T/M/F/N mutation selected
in vitro or in clinical studies show reduced susceptibility to baloxavir. Baloxavir is active against neuraminidase inhibitor resistant strains including H274Y in A/H1N1, E119V and R292K in A/H3N2, and R152K and D198E in type B virus, H274Y in A/H5N1, R292K in A/H7N9.
The relationship between antiviral activity in cell culture and inhibition of influenza virus replication in humans has not been established.
At twice the expected exposure from recommended dosing, XOFLUZA did not prolong the QTc interval.
Clinical/Efficacy Studies: Otherwise healthy patients: Study 1601T0831: Study 1601T0831 is a randomized, double-blind, multicenter, placebo- and active-controlled study designed to evaluate the efficacy and safety of single oral dose of XOFLUZA compared with placebo or oseltamivir in otherwise healthy adult and adolescent patients (aged ≥12 years to ≤64 years) with influenza.
A total of 1436 patients were randomized to receive treatment in the 2016-2017 Northern Hemisphere influenza season. Patients were randomized to receive 40 mg or 80 mg of XOFLUZA according to weight (<80kg or ≥80kg respectively), oseltamivir 75 mg twice daily for 5 days (if aged >20 years) or placebo. The predominant influenza virus strain in this study was the A/H3 subtype (84.8% to 88.1%) followed by the B type (8.3% to 9.0%) and the A/H1N1pdm subtype (0.5% to 3.0%). The primary efficacy endpoint was time to alleviation of symptoms (cough, sore throat, headache, nasal congestion, feverishness or chills, muscle or joint pain, and fatigue). A statistically significant and clinically meaningful improvement in the primary endpoint was seen for XOFLUZA when compared with placebo, see Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
When the XOFLUZA group was compared to the oseltamivir group, there was no statistically significant difference in time to alleviation of symptoms (53.5 h vs 53.8 h respectively), see Table 2.
Click on icon to see table/diagram/image
Secondary endpoints included time to resolution of fever and culture-based assessment of time to cessation of viral shedding (by virus titer).
Resolution of Fever: Following study drug administration there was faster resolution of fever in the XOFLUZA group compared with the placebo group. The median time to resolution of fever in patients treated with XOFLUZA was 24.5 hours (95% CI: 22.6, 26.6) compared with 42.0 hours (95% CI: 37.4, 44.6) in those receiving placebo. No difference was noted in duration of fever in the Xofluza group compared with the oseltamivir group.
Antiviral Activity: Patients treated with XOFLUZA showed a rapid reduction in virus titer. The median time to cessation of viral shedding determined by virus titer was 24.0 hours (95% CI: 24.0, 48.0) in the XOFLUZA group compared with 72.0 hours (95% CI: 72.0, 96.0) in the oseltamivir group and 96.0 hours (95% CI: 96.0, 96.0) in the placebo group.
Study 1518T0821: The phase 2 study was designed to evaluate the efficacy and safety of a single oral dose of XOFLUZA compared with placebo in otherwise healthy adult patients (aged ≥20 years to ≤64 years) with influenza. A total of 400 patients were randomized to one of three dose groups of XOFLUZA (10, 20 or 40 mg) or placebo in the 2015-2016 Northern Hemisphere influenza season in Japan. The predominant influenza virus strain was A/H1N1pdm subtype (61% to 71%) followed by B subtype (21% to 24%) and A/H3N2 subtype (5% to 13%).
The median time to alleviation of symptoms was significantly shorter (p<0.05) compared with placebo in all dose groups . At 40 mg the median time to alleviation of symptoms was 49.5 hours (95% CI: 44.5, 64.4) in the group versus 77.7 hours (95% CI: 67.6, 88.7) in the placebo group.
Resolution of Fever: The median time to resolution of fever was significantly reduced in all dose groups compared with placebo. At 40 mg the median time was 28.9 hours (95% CI: 24.5, 34.7) versus 45.3 hours (95% CI: 35.6, 54.0) in the placebo group. Viral endpoint results were consistent with those in study 1601T0831.
High risk patients: Study 1602T0832: Study 1602T0832 is a randomized, double-blind, multicenter, placebo- and active-controlled study designed to evaluate the efficacy and safety of single oral dose of XOFLUZA compared with placebo or oseltamivir in adult and adolescent patients (aged ≥12 years) with influenza at high risk of influenza complications (e.g. asthma or chronic lung disease, endocrine disorders, heart disease, age ≥ 65 years, metabolic disorders, morbid obesity).
A total of 2184 patients were randomized to receive a single oral dose of 40 mg or 80 mg of XOFLUZA according to body weight (patients who weighed 40 to < 80 kg received 40 mg and patients who weighed ≥ 80 kg received 80 mg), oseltamivir 75 mg twice daily for 5 days, or placebo. The predominant influenza viruses in this study were the A/H3 subtype (46.9% to 48.8%) and influenza B (38.3% to 43.5%). The primary efficacy endpoint was time to improvement of influenza symptoms (cough, sore throat, headache, nasal congestion, feverishness or chills, muscle or joint pain, and fatigue). A statistically significant improvement in the primary endpoint was observed for XOFLUZA when compared with placebo, see Table 3.
Click on icon to see table/diagram/image
When the XOFLUZA group was compared to the oseltamivir
group, there was no statistically significant difference in time to improvement
of influenza symptoms (73.2 h vs 81.0 h respectively), see Table 4.
Click on icon to see table/diagram/image
Virus Subtype: For patients infected with type A/H3 virus (predominant strain), the median time to improvement of influenza symptoms was shorter in the XOFLUZA group compared with the placebo group but not when compared with the oseltamivir group (see Table 5). In the subgroup of patients infected with type B virus, the median time to improvement of influenza symptoms was shorter in the XOFLUZA group compared with both the placebo and oseltamivir group. (See Table 5.)
Click on icon to see table/diagram/image
Resolution of Fever: The proportion of patients who had fever was reduced more rapidly in the XOFLUZA group than in the placebo group following study drug administration. The median time to resolution of fever was 30.8 hours (95% CI: 28.2, 35.4) in the XOFLUZA group compared with 50.7 hours (95% CI: 44.6, 58.8) in the placebo group. No clear differences between the XOFLUZA group and the oseltamivir group were observed.
Incidence of Influenza-Related Complications: The overall incidence of influenza-related complications (death, hospitalization, sinusitis, otitis media, bronchitis, and/or pneumonia) was 2.8% (11/388 patients) in the XOFLUZA group compared with 10.4% (40/386 patients) in the placebo group and 4.6% (18/389 patients) in the oseltamivir group. The lower overall incidence of influenza-related complications in the baloxavir marboxil group compared with the placebo group was mainly driven by lower incidences of bronchitis (1.8% vs. 6.0%, respectively) and sinusitis (0.3% vs. 2.1%, respectively).
The proportion of patients requiring systemic antibiotics for infections secondary to influenza infection was lower in the XOFLUZA group (3.4%) compared with the placebo group (7.5%), and the difference between these 2 groups was statistically significant (p=0.0112). The proportion of patients requiring systemic antibiotics in the XOFLUZA group was comparable with the proportion in the oseltamivir group (3.9%).
Antiviral Activity: Patients at high risk of influenza complications, treated with XOFLUZA, showed a rapid reduction in virus titer and a significantly shortened time to cessation of viral shedding. The median time to cessation of viral shedding determined by virus titer was 48 hours in the XOFLUZA group compared with 96 hours in the placebo group and the oseltamivir group.
Resistance Monitoring during Clinical Development: Influenza A virus isolates with treatment-emergent amino acid substitutions in the PA protein at position I38T/F/M/N associated with > 10 fold reduced susceptibility to baloxavir and influenza B virus isolates with treatment-emergent amino acid substitutions in the PA protein at position I38T associated with > 5 fold reduced susceptibility to baloxavir were observed in clinical studies. The clinical impact of this reduced susceptibility is unknown.
No pre-treatment isolates, with amino acid substitutions associated with reduced susceptibility to baloxavir, were found in the clinical studies.Prescribers should consider available information from the CDC on influenza virus drug susceptibility patterns and treatment effects when deciding whether to use XOFLUZA.
In the phase 3 study in otherwise healthy patients (1601T0831), PA/I38T/M were detected in 36 of 370 patients (9.7%) in the XOFLUZA treatment group. In the phase 3 study in high risk patients (1602T0832), PA/I38T/M/N were detected in 15 of 290 patients (5.2%) in the XOFLUZA treatment group.
Cross Resistance: No single amino acid substitution has been identified that could confer cross-resistance between baloxavir and neuraminidase inhibitors (e.g., peramivir, oseltamivir, zanamivir). However, a virus may carry amino acid substitutions associated with reduced susceptibility to baloxavir in the PA protein and to neuraminidase inhibitors in the neuraminidase and may therefore exhibit reduced susceptibility to both classes of inhibitors. The clinical relevance of phenotypic cross resistance evaluations has not been established.
Immunogenicity: Immune Response: Interaction studies with influenza vaccines and baloxavir marboxil have not been conducted. In studies of naturally acquired and experimental influenza, treatment with XOFLUZA did not impair normal humoral antibody response to infection.
Pharmacokinetics: After oral administration baloxavir marboxil is extensively converted to its active metabolite, baloxavir, predominantly by arylacetamide deacetylase in the gastrointestinal lumen, intestinal epithelium, and liver. The plasma concentration of baloxavir marboxil was very low or below the limit of quantitation (<0.100 ng/mL).
The pharmacokinetic parameters of baloxavir in Japanese healthy adult subjects after a single oral administration of 40 mg baloxavir marboxil in the fasted and fed states are summarized in Table 6. The pharmacokinetic parameters of baloxavir in Caucasian healthy adult subjects after a single oral administration of 80 mg baloxavir marboxil in the fasted state are summarized in Table 7. (See Tables 6 and 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Absorption: Following a single oral administration of 80 mg of baloxavir marboxil, the time to achieve peak plasma concentration (T
max) of baloxavir was reached at approximately 4 hours in the fasted state. The absolute bioavailability of baloxavir marboxil has not been established.
Food effect: A food-effect study involving administration of baloxavir marboxil to healthy volunteers under fasting conditions and with a meal (approximately 400 to 500 kcal including 150 kcal from fat) indicated that the Cmax and AUC of baloxavir were decreased by 48% and 36%, respectively, under fed conditions. Tmax was unchanged in the presence of food. In clinical studies with influenza patients where XOFLUZA was administered with or without food, no clinically relevant differences in efficacy were observed.
Distribution: In an
in vitro study, the binding of baloxavir to human serum proteins, primarily albumin, is 92.9% to 93.9%. The apparent volume of distribution of baloxavir following a single oral administration of 80 mg of baloxavir marboxil is approximately 1180 liters in Caucasian patients and 647 liters in Japanese subjects.
Metabolism: In vitro studies revealed that arylacetamide deacetylase in the gastrointestinal lumen, intestinal epithelium, and the liver mainly contributes to the conversion from baloxavir marboxil to baloxavir and baloxavir is primarily metabolized by UGT1A3 with minor contribution from CYP3A4.
In the human mass balance study, after administration of a single oral dose of 40 mg of [
14C]-labeled baloxavir marboxil, baloxavir accounted for 82.2% of the plasma AUC for total radioactivity. Baloxavir glucuronide (16.4% of the plasma AUC for total radioactivity) and (12aR,5R,11S) sulfoxide of baloxavir (1.5% of the plasma AUC for total radioactivity) were also detected in plasma, confirming that the
in vivo metabolism of baloxavir marboxil occurs via ester hydrolysis to form baloxavir with subsequent metabolism of baloxavir to form sulfoxides, and a glucuronide.
Excretion:
Baloxavir marboxil and baloxavir were excreted mainly via fecal route in humans. Following a single oral administration of 40 mg of [
14C]-labeled baloxavir marboxil, the amount of total radioactivity excreted were 80.1% of the administered dose in the feces and 14.7% in urine. The amount of baloxavir excreted in the urine was 3.3% of the administered dose.
Elimination: The apparent terminal elimination half-life (t
½,z) of baloxavir after a single oral administration of baloxavir marboxil is 79.1 hours in Caucasian patients, and 93.9 hours in Japanese subjects, see Tables 6 and 7.
Linearity/non-linearity: Following single oral administration of baloxavir marboxil, baloxavir exhibits linear pharmacokinetics in the fasted state within the dose range of 6 mg to 80 mg.
Pharmacokinetics in Special Populations: Body weight: Body weight is identified as the significant covariate based on the population pharmacokinetic analysis. The dose proposed in adults is 40 mg for patients with body weight 40 kg to <80 kg, and 80 mg for patients with body weight ≥80 kg.
Gender: A population pharmacokinetic analysis did not identify a clinically meaningful effect of gender on the pharmacokinetics of baloxavir. No dose adjustment based on gender is required.
Race: Based on a population pharmacokinetic analysis, race is a covariate on CL/F of baloxavir in addition to body weight, however, no dose adjustment of baloxavir marboxil based on race is required.
Age: A population pharmacokinetic analysis using plasma baloxavir concentrations from clinical studies with baloxavir marboxil for subjects aged 12 to 64 years did not identify a clinically meaningful effect of age on the pharmacokinetics of baloxavir.
Pediatric Population: The pharmacokinetics of XOFLUZA in pediatric patients (<12 years of age) has not been established.
Geriatric Population: Pharmacokinetic data collected in patients ≥65 years show that drug exposure to baloxavir was similar to patients aged ≥12 to 64 years.
Renal impairment: The effects of renal impairment on the pharmacokinetics of baloxavir marboxil or baloxavir have not been evaluated. Renal impairment is not expected to alter the elimination of baloxavir marboxil or baloxavir. Renal excretion represents a minor pathway of elimination for baloxavir marboxil or baloxavir. A population pharmacokinetic analysis did not identify a clinically meaningful effect of renal function on the pharmacokinetics of baloxavir. No dose adjustment is required in patients with renal impairment.
Baloxavir is unlikely to be significantly removed by dialysis.
Hepatic impairment: Geometric mean ratios (90% confidence interval) of Cmax and AUC of baloxavir in patients with moderate hepatic impairment (Child-Pugh class B) compared to healthy controls were 0.80 (0.50-1.28) and 1.12 (0.78-1.61), respectively. Since no clinically meaningful differences in the pharmacokinetics of baloxavir were observed in patients with moderate hepatic impairment (Child-Pugh class B) compared with healthy controls with normal hepatic function, no dose adjustment is required in patients with mild or moderate hepatic impairment.
The pharmacokinetics in patients with severe hepatic impairment has not been evaluated.
Toxicology: Preclinical safety data: Nonclinical data reveal no special hazards for humans based on conventional studies of safety pharmacology, acute and repeated dose toxicity.
Carcinogenicity: Carcinogenicity studies have not been performed with baloxavir marboxil.
Genotoxicity: The pro-drug baloxavir marboxil, and its active form, baloxavir, were negative in bacterial reverse mutation tests, micronucleus tests with cultured mammalian cells, and baloxavir marboxil was negative in an
in vivo rodent micronucleus test.
Impairment of Fertility: Baloxavir marboxil had no effects on fertility when given orally to male and female rats at doses up to 1000 mg/kg/day, which is equivalent to 5-times the human exposure based on AUC
0-24hr.
Reproductive toxicity: Baloxavir marboxil did not cause malformations in rats or rabbits. The oral embryo-fetal development study of baloxavir marboxil in rats with daily doses from gestation day 6 to 17 revealed no signs of maternal or fetal toxicity up to the highest tested dose of 1000 mg/kg/day, which is equivalent to 5-times the human exposure based on AUC
0-24hr.
In rabbits, a dose level of 1000 mg/kg/day (equivalent to 12-times the human exposure based on AUC
0-24hr following the MHRD) caused maternal toxicity resulting in 2 miscarriages out of 19 and an increased incidence of fetuses with a skeletal variation (cervical rib), but no malformations. This minor skeletal variation is reabsorbed during the growing process of adjacent cervical vertebra. A dose of 100 mg/kg/day (equivalent to 7-times the human exposure based on AUC
0-24hr) in rabbits was without adverse effects.
The pre- and postnatal study in rats did not show drug-related adverse findings in dams and pups up to the highest tested dose of 1000 mg/kg/day, which is equivalent to 5-times the human exposure based on AUC
0-24hr.
Other: Not applicable.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image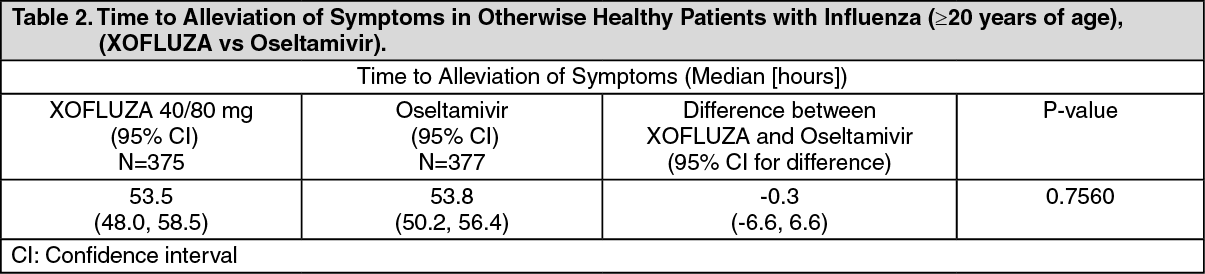 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image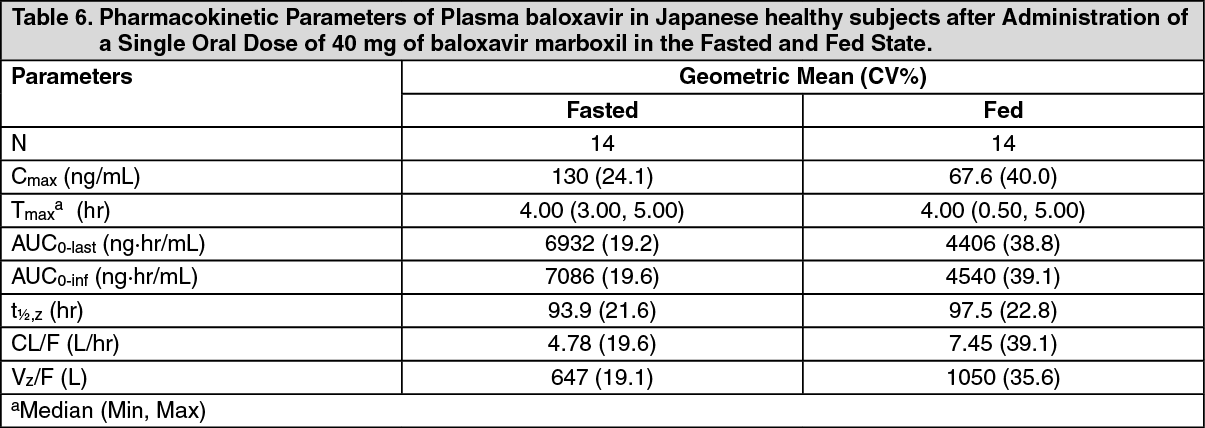 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image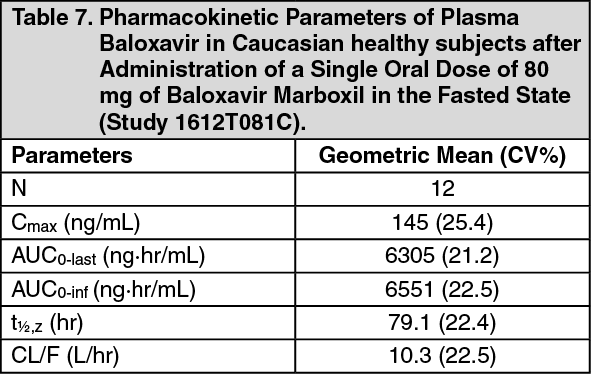 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image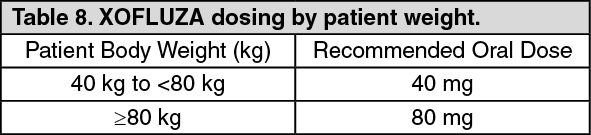 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
