Pharmacotherapeutic Group: Oral direct thrombin inhibitor.
ATC Code: B01AE07.
Pharmacology: Pharmacodynamics: General: Dabigatran etexilate is a small molecule prodrug which does not exhibit any pharmacological activity. After oral administration, dabigatran etexilate is rapidly absorbed and converted to dabigatran by esterase-catalysed hydrolysis in plasma and in the liver. Dabigatran is a potent, competitive, reversible direct thrombin inhibitor and is the main active principle in plasma.
Since thrombin (serine protease) enables the conversion of fibrinogen into fibrin during the coagulation cascade, its inhibition prevents the development of thrombus. Dabigatran also inhibits free thrombin, fibrin-bound thrombin and thrombin-induced platelet aggregation.
In vivo and
ex vivo animal studies have demonstrated antithrombotic efficacy and anticoagulant activity of dabigatran after IV administration and of dabigatran etexilate after oral administration in various animal models of thrombosis.
There is a close correlation between plasma dabigatran concentrations and degree of anticoagulant effect.
Dabigatran prolongs the activated partial thromboplastin time (aPTT), ecarin clotting time (ECT) and thrombin time (TT).
Clinical Trials in Primary Venous Thromboembolism (VTE) Prevention Following Major Joint Replacement Surgery: In 2 large, randomized, parallel group, double-blind, dose-confirmatory trials, patients undergoing elective major orthopaedic surgery (1 for knee replacement surgery and 1 for hip replacement surgery) received dabigatran etexilate 75 mg or 110 mg within 1-4 hrs of surgery followed by 150 mg or 220 mg once daily thereafter, haemostasis having been secured, or enoxaparin 40 mg on the day prior to surgery and once daily thereafter.
In the RE-MODEL trial (knee replacement), treatment was for 6-10 days and in the RE-NOVATE trial (hip replacement), for 28-35 days. A total of 2,076 patients (knee) and 3,494 (hip) were treated, respectively.
The results of the knee study (RE-MODEL) with respect to the primary endpoint, total including asymptomatic VTE plus all-cause mortality showed that the antithrombotic effect of both doses of dabigatran etexilate were statistically non-inferior to that of enoxaparin.
Similarly, total including asymptomatic VTE and all-cause mortality constituted the primary endpoint for the hip study (RE-NOVATE). Again, dabigatran etexilate at both once-daily doses was statistically non-inferior to enoxaparin 40 mg daily.
Furthermore, in a 3rd randomized, parallel group, double-blind trial (RE-MOBILIZE), patients undergoing elective total knee surgery received dabigatran etexilate 75 mg or 110 mg within 6-12 hrs of surgery followed by 150 mg and 220 mg once daily thereafter. The treatment duration was 12-15 days. In total, 2,615 patients were randomized and 2,596 were treated. The comparator dosage of enoxaparin was 30 mg twice daily according to the US label. In the RE-MOBILIZE trial, non-inferiority was not established. There were no statistical differences in bleeding between the comparators.
In addition, a randomized, parallel group, double-blind, placebo-controlled phase II study in Japanese patients where dabigatran etexilate 110 mg, 150 mg and 220 mg was administered at the next day after elective total knee replacement surgery was evaluated. The Japanese study showed a clear dose-response relationship for the efficacy of dabigatran etexilate and a placebo-like bleeding profile.
In RE-MODEL and RE-NOVATE, the randomization to the respective study medication was done pre-surgery, and in the RE-MOBILIZE and the Japanese placebo-controlled trial, the randomization to the respective study medication was done post-surgery. This is of note especially in the safety evaluation of these trials. For this reason, the trials are grouped in pre- and post-surgery randomized trials in Table 1.
Data for the major VTE and VTE-related mortality endpoint and adjudicated major bleeding endpoints are shown in Table 1 as follows. (See Table 1.)
Venous thromboembolism was defined as the composite incidence of deep vein thrombosis and pulmonary embolism.
 Click on icon to see table/diagram/image
Clinical Trials in Prevention of Stroke and Systemic Embolism in Patients with Atrial Fibrillation (AF):
Click on icon to see table/diagram/image
Clinical Trials in Prevention of Stroke and Systemic Embolism in Patients with Atrial Fibrillation (AF): The clinical evidence for the efficacy of dabigatran etexilate is derived from the RE-LY (Randomized Evaluation of Long-Term Anticoagulant Therapy) study, a multicenter, multinational, randomized, parallel group study of 2 blinded doses of dabigatran etexilate (110 mg and 150 mg twice daily) compared to open-label warfarin in patients with AF at moderate to high risk of stroke or systemic embolism. The primary objective in this study was to determine if dabigatran was non-inferior to warfarin in reducing the occurrence of the composite endpoint, stroke and systemic embolic events (SEE).
In the RE-LY study, a total of 18,113 patients were randomized, with a mean age of 71.5 years and a mean CHADS
2 score of 2.1. The population had approximately equal proportions of patients with CHADS
2 score 1, 2 and ≥3. The patient population was 64% male, 70% Caucasian and 16% Asian. RE-LY had a median treatment of 20 months with dabigatran etexilate given as fixed dose without coagulation monitoring. In addition to documented non-valvular AF (eg, persistent AF or paroxysmal), patients had one of the following additional risk factors for stroke: Previous stroke, transient ischemic attack or systemic embolism; left ventricular ejection fraction <40%; symptomatic heart failure, ≥ New York Heart Association (NYHA) Class 2; age ≥75 years; age ≥65 years associated with one of the following: Diabetes mellitus, coronary artery disease (CAD) or hypertension.
The concomitant diseases of patients in this trial included hypertension 79%, diabetes 23% and CAD 28%. Fifty percent (50%) of the patient population was vitamin K antagonist (VKA)-naive defined as <2 months total lifetime exposure. Thirty-two percent (32%) of the population had never been exposed to a VKA. For those patients randomized to warfarin, the time in therapeutic range (INR 2-3) for the trial was a median of 67%.
Concomitant medications included aspirin (ASA) (25% of the subjects used at least 50% of the time in study), clopidogrel (3.6%), ASA + clopidogrel (2%), nonsteroidal anti-inflammatory drugs (NSAIDs) (6.3%), β-blockers (63.4%), diuretics (53.9%), statins (46.4%), angiotensin-converting enzyme (ACE) inhibitors (44.6%), angiotensin-receptor blockers (26.1%), oral hypoglycemics (17.5%), insulin (5.2%), digoxin (29.4%), amiodarone (11.3%), diltiazem (8.9%), verapamil (5.4%) and proton-pump inhibitors (PPIs) (17.8%).
For the primary endpoint, stroke and systemic embolism, no subgroups (ie, age, weight, gender, renal function, ethnicity) were identified with a different risk-ratio compared to warfarin.
This study demonstrated that dabigatran etexilate, at a dose of 110 mg twice daily, is non-inferior to warfarin in the prevention of stroke and systemic embolism in subjects with AF, with a reduced risk of intracranial hemorrhage and total bleeding. The higher dose of 150 mg twice daily significantly reduces the risk of ischemic and hemorrhagic stroke, vascular death, intracranial hemorrhage and total bleeding compared to warfarin. The lower dose of dabigatran has a significantly lower risk of major bleeding compared to warfarin (see Figure 1 and Tables 2-6).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The net clinical benefit (NCB) as measured by the unweighted composite clinical endpoint of stroke, systemic embolism, pulmonary embolism, acute myocardial infarction, vascular deaths and major bleeds was assessed and is presented as part of Table 5. The yearly event rates for the dabigatran etexilate groups were lower compared to the warfarin group. The risk reduction for this composite endpoint was 8% and 10% for the dabigatran etexilate 110 mg and 150 mg twice daily treatment groups.
Other components evaluated included all hospitalizations which had statistically significant fewer hospitalizations at dabigatran etexilate 110 mg twice daily compared to warfarin (7% risk reduction, 95% CI 0.87, 0.99, p=0.021).
Click on icon to see table/diagram/image
In the RE-LY study, potential abnormalities of liver function tests (LFTs) occurred with a comparable or lower incidence in dabigatran etexilate versus warfarin-treated patients. (See Table 6.)
Click on icon to see table/diagram/image
Clinical Trials for the Prevention of Thromboembolism in Patients With Prosthetic Heart Valves: A phase II study examined dabigatran etexilate and warfarin in a total of 252 patients with recent mechanical heart valve replacement surgery (ie, within the current hospital stay) and in patients who received a mechanical heart valve replacement >3 months ago. An imbalance in thromboembolic and total (mainly minor) bleeding events in disfavour of dabigatran etexilate was observed in this trial.
In the early postoperative patients, major bleeding manifested predominantly as haemorrhagic pericardial effusions, specifically in patients who started dabigatran etexilate early (ie, on day 3) after heart valve replacement surgery.
Clinical Trials in Treatment of Acute Deep Vein Thrombosis (DVT) and/or Pulmonary Embolism (PE) and Prevention of Related Death: Clinical evidence has demonstrated dabigatran etexilate to be an effective and safe treatment for DVT and/or PE in 2 multicenter, randomised, double-blind, parallel group, replicate studies RE-COVER and RE-COVER II. These studies compared dabigatran etexilate (150 mg twice daily) with warfarin (target INR 2-3) in patients with acute DVT and/or PE. The primary objective of these studies was to determine if dabigatran was non-inferior to warfarin in reducing the occurrence of the primary endpoint which was the composite of recurrent symptomatic DVT and/or PE and related deaths within the 6 month acute treatment period.
In the pooled RE-COVER and RE-COVER II studies, a total of 5,153 patients were randomized and 5,107 were treated. The index events at baseline: DVT-68.5%, PE-22.2%, PE and DVT-9.1%. The most frequent risk factors were history of DVT and/or PE-21.5%, surgery/trauma-18.1%, venous insufficiency-17.6%, and prolonged immobilisation-14.6%. Patients’ baseline characteristics: Mean age was 54.8 years, males 59.5%, Caucasian 86.1%, Asian 11.8%, Blacks 2.1%. The co-morbidities included: Hypertension 35.5%, diabetes mellitus 9%, CAD 6.8% and gastric or duodenal ulcer 4.1%.
The duration of treatment with fixed-dose of dabigatran was 174 days without coagulation monitoring. For patients randomized to warfarin, the median time in therapeutic range (INR 2-3) was 60.6%. Concomitant medications included vasodilators 28.5%, agents acting on the renin-angiotensin system 24.7%, lipid-lowering agents 19.1%, β-blockers 14.8%, calcium channel blockers 9.7%, nonsteroidal anti-inflammatory drugs (NSAIDs) 21.7%, aspirin 9.2%, antiplatelet agents 0.7%, P-gp inhibitors 2% (verapamil - 1.2% and amiodarone - 0.4%).
Two trials in patients presenting with acute DVT and/or PE treated initially for at least 5 days of parenteral therapy, RE-COVER and RE-COVER II, demonstrated that treatment with dabigatran etexilate 150 mg twice daily was non-inferior to the treatment with warfarin (p-values for non-inferiority: RE-COVER p<0.0001, RE-COVER II p=0.0002). Bleeding events (MBEs, MBE/CRBEs and any bleeding) were significantly lower in patients receiving dabigatran etixilate 150 mg twice daily as compared with those receiving warfarin. (See Figure 2 and Table 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Other Measures Evaluated: Treatment of acute deep vein thrombosis (DVT) and/or pulmonary embolism (PE) and prevention of related death: Myocardial infarction occurred at a low frequency in all 4 of the VTE studies for all treatment groups. Cardiac death occurred in 1 patient in the warfarin treatment group.
In the 3 active controlled-studies, a higher rate of myocardial infarction was reported in patients who received dabigatran etexilate (20; 05%) than in those who received warfarin (5; 0.1%).
In the RE-SONATE study, which compared dabigatran etexilate to placebo, there was 1 myocardial infarction event in each of the treatment groups, resulting in myocardial infarction rates with dabigatran equal to myocardial infarction rates with placebo.
Liver Function Tests: Treatment of acute DVT and/or PE and prevention of related death: In the active controlled-studies RE-COVER, RE-COVER II and RE-MEDY, potential abnormalities of liver function tests (LFT) occurred with a comparable or lower incidence in dabigatran etexilate versus warfarin-treated patients. In RE-SONATE, there was no marked difference between the dabigatran- and placebo groups with regard to possible clinically significant abnormal LFT values.
Clinical Trials in Prevention of Recurrent of Deep Vein Thrombosis (DVT) and/or Pulmonary Embolism (PE) and Related Death: Clinical evidence has demonstrated dabigatran etexilate to be an effective and safe treatment for recurrent DVT and/or PE. Two randomized, parallel-group, double-blind studies were performed in patients previously treated with anticoagulation therapy. RE-MEDY, warfarin-controlled study, enrolled patients already treated for 3-12 months with the need for further anticoagulant treatment and RE-SONATE, the placebo-controlled study, enrolled patients already treated for 6-18 months with vitamin K inhibitors.
The objective of the RE-MEDY study was to compare the safety and efficacy of oral dabigatran etexilate (150 mg twice daily) to warfarin (target INR 2-3) for the long-term treatment and prevention of recurrent, symptomatic DVT and/or PE. A total of 2,866 patients were randomized and 2,856 patients were treated. The index events at baseline: DVT - 65.1%, PE - 23.1%, PE and DVT - 11.7%. Patients’ baseline characteristics: Mean age 54.6 years, males 61%, Caucasian 90.1%, Asian 7.9%, Blacks 2%. Co-morbidities included hypertension 38.6%, diabetes mellitus 9%, coronary artery disease (CAD) 7.2% and gastric or duodenal ulcer 3.8%. Concomitant medications: Agents acting on the renin-angiotensin system 27.9%, vasodilators 26.7, lipid-lowering agents 20.6%, NSAIDs 18.3%, β-blockers 16.3%, calcium channel blockers 11.1%, aspirin 7.7%, P-gp inhibitors 2.7% (verapamil 1.2% and amiodarone 0.7%), antiplatelets 0.9%. Duration of dabigatran exilate treatment ranged from 6-36 months (median-534 days). For patients randomized to warfarin, the median time in therapeutic range (INR 2-3) was 64.9%.
RE-MEDY demonstrated that treatment with dabigatran etexilate 150 mg twice daily was non-inferior to warfarin (p=0.0135 for non-inferiority). Bleeding events (MBEs/CRBEs; any bleeding) were significantly lower in patients receiving dabigatran etexilate as compared with those receiving warfarin.
As in the pooled RE-COVER/RE-COVER II studies, in RE-MEDY concomitant use of P-gp inhibitors was reported by few patients (2.7%); verapamil (1.2%) and amiodarone (0.7%) were the most frequent. In the pooled acute VTE treatment studies, concomitant use of P-gp inhibitors was reported by few patients (2%); most frequent were verapamil (1.2% overall) and amiodarone (0.4% overall). (See Figure 3 and Table 8.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The objective of the RE-SONATE study was to evaluate superiority of dabigatran etexilate versus placebo for the prevention of recurrent symptomatic DVT and/or PE in patients who had already completed 6-18 months of treatment with VKA. The intended therapy was 6 months dabigatran etexilate 150 mg twice daily without need for monitoring.
The index events at baseline: DVT 64.5%, PE 27.8%, PE and DVT 7.7%. A total of 1,353 patients were randomized and 1,343 patients treated. Patients’ baseline characteristics: Mean age 55.8 years, males 55.5%, Caucasian 89%, Asian 9.3%, Blacks 1.7%. Co-morbidities included hypertension 38.8%, diabetes mellitus (DM) 8%, coronary artery disease (CAD) 6% and gastric or duodenal ulcer 4.5%. Concomitant medications: Agents acting on the renin-angiotensin system 28.7%, vasodilators 19.4%, lipid-lowering
agents 17.9%, β-blockers 18.5%, calcium channel blockers 8.9%, NSAIDs 12.1%, aspirin 8.3%, antiplatelets 0.7% and P-gp inhibitors 1.7% (verapamil 1% and amiodarone 0.3%).
RE-SONATE demonstrated dabigatran etexilate was superior to placebo for the prevention of recurrent symptomatic DVT/PE events including unexplained deaths, with a risk reduction of 92% during the treatment period (p<0.0001). All secondary and sensitivity analyses of the primary endpoint and all secondary endpoints showed superiority of dabigatran etexilate over placebo. The rates of MBEs and the combination of MBEs/CRBEs were significantly higher in patients receiving dabigatran etexilate as compared with those receiving placebo.
The study included observational follow-up for 12 months after the conclusion of treatment. After discontinuation of study medication the effect was maintained until the end of the follow-up, indicating that the initial treatment effect of dabigatran etexilate was sustained. No rebound effect was observed. At the end of the follow-up, VTE events in patients treated with dabigatran etexilate was 6.9% versus 10.7% among the placebo group [hazard ratio 0.61 (0.42, 0.88), p=0.0082]. (See Figure 4 and Table 9.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Other Measures Evaluated:
Prevention of recurrent DVT and/or PE and Related death: Myocardial infarction occurred at a low frequency in all 4 of the VTE studies for all treatment groups. Cardiac death occurred in 1 patient in the warfarin-treatment group. In the 3 active controlled-studies, a higher rate of myocardial infarction was reported in patients who received dabigatran etexilate (20; 0.5%) than in those who received warfarin (5; 0.1%).
In the RE-SONATE study, which compared dabigatran etexilate to placebo, there was 1 myocardial infarction event in each of the treatment groups, resulting in myocardial infarction rates with dabigatran equal to myocardial infarction rates with placebo.
Liver Function Tests: Prevention of recurrent DVT and/or PE and related death: In the active controlled-studies RE-COVER, RE-COVER II and RE-MEDY, potential abnormalities of liver function tests (LFT) occurred with a comparable or lower incidence in dabigatran etexilate versus warfarin-treated patients. In RE-SONATE, there was no marked difference between the dabigatran- and placebo groups with regard to possible clinically significant abnormal LFT values.
Pharmacokinetics: After oral administration of dabigatran etexilate in healthy volunteers, the pharmacokinetic profile of dabigatran in plasma is characterized by a rapid increase in plasma concentrations with peak concentration (C
max) attained within 0.5 and 2 hrs post-administration.
Peak plasma concentration (C
max) and the area under the plasma concentration-time curve (AUC) were dose-proportional. After C
max, plasma concentrations of dabigatran showed a biexponential decline with a mean terminal half-life (t
½) of approximately 11 hrs in healthy elderly subjects. After multiple doses, a terminal t
½ of about 12-14 hrs was observed. The t
½ was independent of dose. However, half-life is prolonged if renal function is impaired as shown below (see Table 10).
Click on icon to see table/diagram/image
The absolute bioavailability of dabigatran following oral administration of dabigatran etexilate as HPMC capsule was approximately 6.5%.
Food does not affect the bioavailability of dabigatran etexilate but delays the time-to-peak plasma concentrations by 2 hrs.
The oral bioavailability may be increased by about 1.8-fold (+75%) compared to the reference capsule formulation when the pellets are taken without the HPMC capsule shell. Hence, the integrity of the HPMC capsules should always be preserved in clinical use to avoid unintentionally increased bioavailability of dabigatran etexilate. Therefore, patients should be advised not to open the capsules and taking the pellets alone (eg, sprinkled over food or into beverages). (See Dosage & Administration.)
A study evaluating postoperative absorption of dabigatran etexilate, 1-3 hrs following surgery, demonstrated relatively slow absorption compared with that in healthy volunteers, showing a smooth plasma concentration-time profile without high C
max. Peak plasma concentrations are reached at 6 hrs following administration, or at 7-9 hrs following surgery (BISTRO Ib). It is noted, however, that contributing factors eg, anesthesia, gastrointestinal paresis and surgical effects will mean that a proportion of patients will experience absorption delay independent of the oral drug formulation. Although this study did not predict whether impaired absorption persists with subsequent doses, it was demonstrated in a further study that slow and delayed absorption is usually only present on the day of surgery. On subsequent days, the absorption of dabigatran is rapid with C
max attained 2 hrs after drug administration.
Metabolism and excretion of dabigatran were studied following a single IV dose of radiolabeled dabigatran in healthy male subjects. After an IV dose, the dabigatran-derived radioactivity was eliminated primarily in the urine (85%). Faecal excretion accounted for 6% of the administered dose. Recovery of the total radioactivity ranged from 88-94% of the administered dose by 168 hrs post-dose.
After oral administration, dabigatran etexilate is rapidly and completely converted to dabigatran, which is the active form in plasma. The cleavage of the prodrug dabigatran etexilate by esterase-catalysed hydrolysis to the active principle dabigatran is the predominant metabolic reaction. Dabigatran is subject to conjugation, forming pharmacologically active acylglucuronides. Four (4) positional isomers, 1-O, 2-O, 3-O, 4-O-acylglucuronide exist, each accounts for <10% of total dabigatran in plasma. Traces of other metabolites were only detectable with highly sensitive analytical methods. Dabigatran is eliminated primarily in the unchanged form in the urine, at a rate of approximately 100 mL/min corresponding to the glomerular filtration rate.
Low (34-35%) concentration-independent binding of dabigatran to human plasma proteins was observed. The volume of distribution of 60-70 L of dabigatran exceeded the volume of total body water indicating moderate tissue distribution of dabigatran.
Special Populations: Renal Impairment: The exposure (AUC) of dabigatran after the oral administration of dabigatran etexilate in a phase I study was approximately 3-fold higher in volunteers with moderate renal insufficiency [creatinine clearance (CrCl) between 30-50 mL/min] than those without renal insufficiency.
In a small number of volunteers with severe renal insufficiency (CrCl 10-30 mL/min), the exposure (AUC) to dabigatran was approximately 6 times higher and the t
½ approximately 2 times longer than that observed in a population without renal insufficiency (see Dosage & Administration and Contraindications).
Clearance of dabigatran by hemodialysis was investigated in patients with end-stage renal disease (ESRD) without atrial fibrillation. Dialysis was conducted with 700 mL/min dialysate flow rate, 4-hr duration, a blood flow rate of either 200 mL/min or 350-390 mL/min. This resulted in a removal of 50% or 60% of free or total dabigatran concentrations, respectively. The amount of drug cleared by dialysis is proportional to the blood flow rate. The anticoagulant activity of dabigatran decreased with decreasing plasma concentrations and the pharmacokinetic/pharmacodynamic (PK/PD) relationship was not affected by the procedure.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial fibrillation: The median CrCl in RE-LY was 68.4 mL/min. Almost ½ (45.8%) of the RE-LY patients had a CrCl >50 to <80 mL/min. Patients with moderate renal impairment (CrCl between 30-50 mL/min) had, on average, 2.29- and 1.8-fold higher pre- and post-dose dabigatran plasma concentrations, respectively, when compared with patients without renal impairment (CrCl ≥80 mL/min).
Treatment of Acute Deep Vein Thrombosis (DVT) and/or Pulmonary Embolism (PE) and Prevention of Related Death: The median CrCl in the RE-COVER study was 100.4 mL/min. Twenty-one and seven tenths percent (21.7%) of patients had mild renal impairment (CrCl >50 to <80 mL/min) and 4.5% of patients had a moderate renal impairment (CrCl between 30-50 mL/min). Patients with mild and moderate renal impairment had on average 1.8-fold and 3.6-fold higher steady-state dabigatran trough concentrations compared with patients with CrCl >80 mL/min. Similar values for CrCl were found in RE-COVER II.
Prevention of Recurrent DVT and/or PE and Related Death: The median CrCl in the RE-MEDY and RE-SONATE studies were 99 mL/min and 99.7 mL/min, respectively. Twenty-two and nine tenths percent (22.9%) and 22.5% of the patients had a CrCl >50 to <80 mL/min, and 4.1% and 4.8% had a CrCl between 30-50 mL/min in in the RE-MEDY and RE-SONATE studies.
Elderly: Specific pharmacokinetic studies with elderly subjects in phase I studies showed an increase of 1.4- to 1.6-fold (+40-60%) in the AUC and of >1.25-fold (+25%) in C
max compared to young subjects.
The AUC
τ,ss and C
max,ss in male and female elderly subjects (>65 yrs) were approximately 1.9- and 1.6-fold higher for elderly females compared to young females, and 2.2- and 2-fold higher for elderly males than in male subjects of 18-40 years.
The observed increase of dabigatran exposure correlated with the age-related reduction in creatinine clearance.
The effect by age on exposure to dabigatran was confirmed in the RE-LY study with about 1.3-fold (+31%) higher trough concentration for subjects ≥75 years, and by about 22% lower trough level for subjects <65 years compared to subjects of age between 65-75 years.
Hepatic Insufficiency: No change in dabigatran exposure was seen in 12 subjects in a phase I study with moderate hepatic insufficiency (Child Pugh B) compared to 12 controls.
Prevention of VTE Events in Patients Who Have Undergone Major Orthopaedic Surgery: Patients with moderate and severe hepatic impairment (Child-Pugh classification B and C) or liver disease expected to have any impact on survival or with elevated liver enzymes ≥2 upper limit of normal (ULN) were excluded in clinical trials.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: Patients with active liver disease including but not limited to the persistent elevation of liver enzymes ≥2 ULN or hepatitis A, B or C were excluded in clinical trials.
Treatment of Acute DVT and/or PE and Prevention of Related Death: Patients with moderate and severe hepatic impairment (Child-Pugh classification B and C) or liver disease expected to have any impact on survival or with elevated liver enzymes ≥2 ULN were excluded in clinical trials.
Prevention of Recurrent DVT and/or PE and Related Death: Patients with moderate and severe hepatic impairment (Child-Pugh classification B and C) or liver disease expected to have any impact on survival or with elevated liver enzymes ≥2 ULN were excluded in clinical trials.
Body Weight: The dabigatran trough concentrations were about 20% lower in patients with a body weight >100 kg compared with 50-100 kg. The majority (80.8%) of the subjects were in the ≥50 kg and <100 kg category, with no clear difference detected. Limited data in patients ≤50 kg are available.
Gender: Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: In atrial fibrillation patients, females had an average 1.3-fold (+30%) higher trough and post-dose concentrations. This finding had no clinical relevance.
Prevention of Venous Thromboembolic Events (VTE) in Patients Who Have Undergone Major Orthopedic Surgery: Drug exposure in the primary VTE prevention studies was about 1.4- to 1.5-fold (+40-50%) higher in female patients. This finding had no clinical relevance.
Ethnic Origin: The pharmacokinetics of dabigatran was investigated in Caucasian and Japanese volunteers after single and multiple doses. Ethnic origin does not affect the pharmacokinetics of dabigatran in a clinically relevant manner.
Limited pharmacokinetic data in Black patients are available which suggest no relevant differences.
Pharmacokinetic Interactions: In vitro interaction studies did not show any inhibition or induction of cytochrome P-450. This has been confirmed by
in vivo studies in healthy volunteers, who did not show any interaction between dabigatran etexilate treatment and the following drugs: Atorvastatin (CYP3A4) and diclofenac (CYP2C9).
Atorvastatin: When dabigatran etexilate was co-administered with atorvastatin, a CYP3A4 substrate, exposure of atorvastatin, atorvastatin metabolites and of dabigatran were unchanged indicating a lack of interaction.
Diclofenac: When dabigatran etexilate was co-administered with diclofenac, a CYP2C9 substrate, pharmacokinetics of both drugs remained unchanged indicating a lack of interaction between dabigatran etexilate and diclofenac.
P-glycoprotein (P-gp) Inhibitor/Inducer Interactions: The prodrug dabigatran etexilate, but not dabigatran, is a substrate of the efflux transporter P-gp. Therefore, co-medications with P-gp transporter inhibitors and inducers have been investigated.
Co-Medication with P-gp Inhibitors: Amiodarone: When dabigatran etexilate was co-administered with a single oral dose of amiodarone 600 mg, the extent and rate of absorption of amiodarone and its active metabolite diethanolamine (DEA) were essentially unchanged. The dabigatran AUC and C
max were increased by about 1.6- and 1.5-fold (+60% and 50%), respectively.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: In the population pharmacokinetics study from RE-LY, no important changes in dabigatran trough levels were observed in patients who received amiodarone (see Interactions).
Dronedarone: When dabigatran etexilate and dronedarone were given at the same time, total dabigatran AUC
0-∞ and C
max values increased by about 2.4- and 2.3-fold (+136% and 125%), respectively, after multiple dosing of dronedarone 400 mg twice daily, and about 2.1- and 1.9-fold (+114% and 87%), respectively, after a single dose of 400 mg. The terminal
t
½ and renal clearance of dabigatran were not affected by dronedarone. When single and multiple doses of dronedarone were given 2 hrs after dabigatran etexilate, the increases in dabigatran AUC
0-∞ were 1.3- and 1.6-fold, respectively.
Verapamil: When dabigatran etexilate was co-administered with oral verapamil, the C
max and AUC of dabigatran were increased depending on timing of administration and formulation of verapamil.
The greatest elevation of dabigatran exposure was observed with the 1st dose of an immediate-release formulation of verapamil administered 1 hr prior to dabigatran etexilate intake [increase of C
max by about 2.8-fold (+180%) and AUC by about 2.5-fold (+150%)]. The effect was progressively decreased with administration of an extended-release formulation [increase of C
max by about 1.9-fold (+90%) and AUC by about 1.7-fold (+70%)] or administration of multiple doses of verapamil [increase of C
max by about 1.6-fold (+60%) and AUC by about 1.5-fold (+50%)]. This can be explained by the induction of P-gp in the gut by chronic verapamil treatment.
There was no meaningful interaction observed when verapamil was given 2 hrs after dabigatran etexilate (increase of C
max by about 10% and AUC by about 20%). This is explained by completed dabigatran absorption after 2 hrs (see Dosage & Administration).
No data are available for the parenteral application of verapamil; based on the mechanism of the interaction, no meaningful interaction is expected.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: In the population pharmacokinetic study from RE-LY, no important changes in dabigatran trough levels were observed in patients who received verapamil (see Interactions).
Ketoconazole: Systemic ketoconazole increased total dabigatran AUC
0-∞ and C
max values by about 2.4-fold (+138% and 135%), respectively, after a single dose of 400 mg and about 2.5-fold (+153% and 149%), respectively, after multiple dosing of ketoconazole 400 mg once daily. The time-to-peak, terminal
t
½ and mean residence time were not affected by ketoconazole.
Clarithromycin: When clarithromycin 500 mg twice daily was administered together with dabigatran etexilate, no clinically relevant pharmacokinetic interaction was observed (increase of C
max by about 15% and AUC by about 19%).
Quinidine: Quinidine was given as a 200-mg dose every 2nd hr up to a total dose of 1000 mg. Dabigatran etexilate was given twice daily over 3 consecutive days, on the 3rd day either with or without quinidine. Dabigatran AUC
τ,ss and C
max,ss were increased, on average, by about 1.5-fold (+53% and 56%), respectively, with concomitant quinidine.
Ticagrelor: When a single dose of dabigatran etexilate 75 mg was co-administered simultaneously with a loading dose of ticagrelor 180 mg, the dabigatran AUC and C
max were increased by 1.73- and 1.95-fold (+73% and 95%), respectively. After multiple doses of ticagrelor 90 mg twice daily, the increase of dabigatran exposure is reduced to 1.56- and 1.46-fold (+56% and 46%) for C
max and AUC, respectively.
Co-Medication with P-gp Substrates: Digoxin: When dabigatran etexilate was co-administered with digoxin, a P-gp substrate, no PK interaction was observed. Neither dabigatran nor the prodrug dabigatran etexilate is a clinically relevant P-gp inhibitor.
Co-Medication with P-gp Inducers: Rifampicin: Pre-dosing of the probe inducer rifampicin at a dose of 600 mg once daily for 7 days decreased total dabigatran peak and total exposure by 65.5% and 67%, respectively. The inducing effect was diminished resulting in dabigatran exposure close to the reference by day 7 after cessation of rifampicin treatment. No further increase in bioavailability was observed after another 7 days.
Co-Medication with Platelet Inhibitors: Acetylsalicylic Acid (ASA): The effect of concomitant administration of dabigatran etexilate and ASA on the risk of bleeding was studied in patients with atrial fibrillation in a phase II study, in which a randomized ASA co-administration was applied. Based on logistic regression analysis, co-administration of ASA and dabigatran etexilate 150 mg twice daily may increase the risk for any bleeding from 12-18% and 24% with ASA 81 mg and 325 mg, respectively.
From the data gathered in the phase III study RE-LY, it was observed that ASA or clopidogrel co-medication with dabigatran etexilate at dosages of 110 mg or 150 mg twice daily may increase the risk of major bleeding. The higher rate of bleeding events by ASA or clopidogrel co-medication was, however, also observed for warfarin.
Nonsteroidal anti-inflammatory drugs (NSAIDs) given for short-term perioperative analgesia have been shown not to be associated with increased bleeding risk when given in conjunction with dabigatran etexilate. There is limited evidence regarding the use of regular NSAID medication with t
½ of <12 hrs during treatment with dabigatran etexilate and this has not suggested additional bleeding risk.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: Nonsteroidal anti-inflammatory drugs increased the risk of bleeding in RE-LY in all treatment groups.
Clopidogrel: In a phase I study in young healthy male volunteers, the concomitant administration of dabigatran etexilate and clopidogrel resulted in no further prolongation of capillary bleeding times (CBT) compared to clopidogrel monotherapy. In addition, dabigatran AUC
τ,ss, C
max,ss and the coagulation measures for dabigatran effect, activated partial thromboplastin time (aPTT), electroconvulsive therapy (ECT) or thrombolytic therapy (TT) (anti-FIIa) or the inhibition of platelet aggregation (IPA) as measure of clopidogrel effect remained essentially unchanged comparing combined treatment and the respective mono-treatments. With a loading dose of clopidogrel 300 mg or 600 mg, dabigatran AUC
τ,ss and C
max,ss were increased by about 1.3- to 1.4-fold (+30-40%) (see previous discussion on ASA).
Antiplatelets or Other Anticoagulants: The concomitant use of dabigatran etexilate and antiplatelets, or other anticoagulants may increase the risk of bleeding (see Precautions).
Co-Medication with Selective Serotonin Reuptake Inhibitors (SSRIs): Selective serotonin reuptake inhibitors (SSRIs) increase the risk of bleeding in RE-LY in all treatment groups.
Co-Medication with Gastric pH-Elevating Agents: The changes in dabigratan exposure determined by population pharmacokinetic analysis caused by proton-pump inhibitors (PPIs) and antacids were not considered clinically relevant because the magnitude of the effect were minor (fractional decrease in bioavailability not significant for antacids and 14.6% for PPIs.
Pantoprazole: When dabigatran etexilate was co-administered with pantoprazole, a decrease in dabigatran AUC of approximately 30% was observed.
Pantoprazole and other PPIs were co-administered with dabigatran etexilate in clinical trials and no effects on bleeding or efficacy were observed.
Prevention of Stroke, Systemic Embolism and Reduction of Vascular Mortality in Patients with Atrial Fibrillation: In the phase III study, RE-LY, PPI co-medication did not result in lower trough levels and on average, only slightly reduced post-dose concentrations (-11%). Accordingly, PPI co-medication seemed to be not associated with a higher incidence of stroke or SEE, especially in comparison with warfarin, and hence, the reduced bioavailability by pantoprazole co-administration seemed to be of no clinical relevance.
Ranitidine: Ranitidine administration, together with dabigatran etexilate, had no meaningful effect on the extent of absorption of dabigatran.
Toxicology: Acute oral toxicity studies were conducted in rats and mice. In both species, the approximate lethal dose after single oral administration was >2000 mg/kg. In dogs and Rhesus monkeys, oral administration of dabigatran etexilate 600 mg/kg did not induce any toxicologically meaningful changes.
In repeat-dose toxicity studies over a maximum of 26 weeks in rats and 52 weeks in Rhesus monkeys, dosages up to 300 mg/kg (free-base equivalent) were used. Generally, these doses were tolerated remarkably well by both rats and Rhesus monkeys. Bleeding problems were observed in association with traumata (eg, blood sampling) within the first 4-6 hrs after administration and are directly related to the pharmacodynamic activity of dabigatran.
Teratology studies were performed with up to 200 mg/kg (free-base equivalent) in rats and rabbits. A slight effect on the morphogenesis of foetuses was observed in rats at 200 mg/kg (free-base equivalent). No teratogenic effects were noted in rabbits.
In the fertility study in rats, no toxicologically remarkable parental findings were noted. With respect to litter parameters, a slight decrease in corpora lutea and an increase in pre-implantation loss led to a decrease in the mean number of implantations in the 200 mg/kg (free-base equivalent) dose group.
Comprehensive
in vitro and
in vivo studies revealed no evidence of a mutagenic potential.
In lifetime toxicology studies in rats and mice, there was no evidence for a tumorigenic potential of dabigatran up to maximum doses of 200 mg/kg (free-base equivalent).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image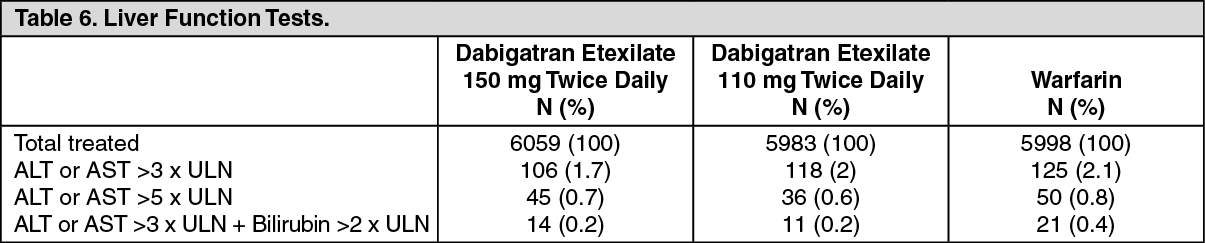 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image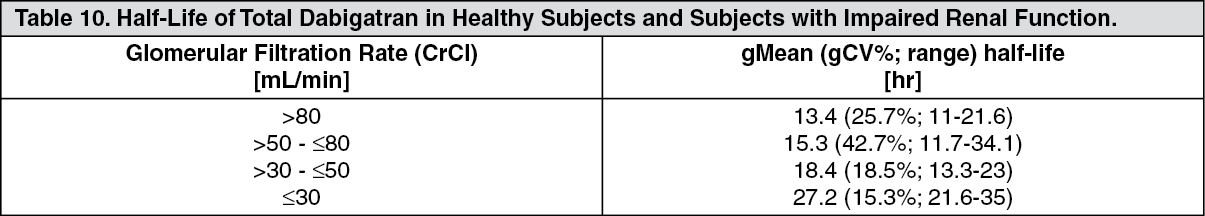 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
