


 Sign Out
Sign Out


Pharmacodynamics: In a study in healthy volunteers (N=10), respiratory rate and oxygen saturation remained within normal limits and there was no evidence of respiratory depression when Dexdor was administered by intravenous infusion at doses within the dose range of 0.2 - 0.7 mcg/kg/hr.
Pharmacokinetics: Following intravenous administration, dexmedetomidine exhibits the following pharmacokinetic parameters: a rapid distribution phase with a distribution half-life (t½) of approximately 6 minutes; a terminal elimination half-life (t½) of approximately 2 hours; and steady-state volume of distribution (Vss) of approximately 118 liters. Clearance is estimated to be approximately 39 L/h. The mean body weight associated with this clearance estimate was 72 kg.
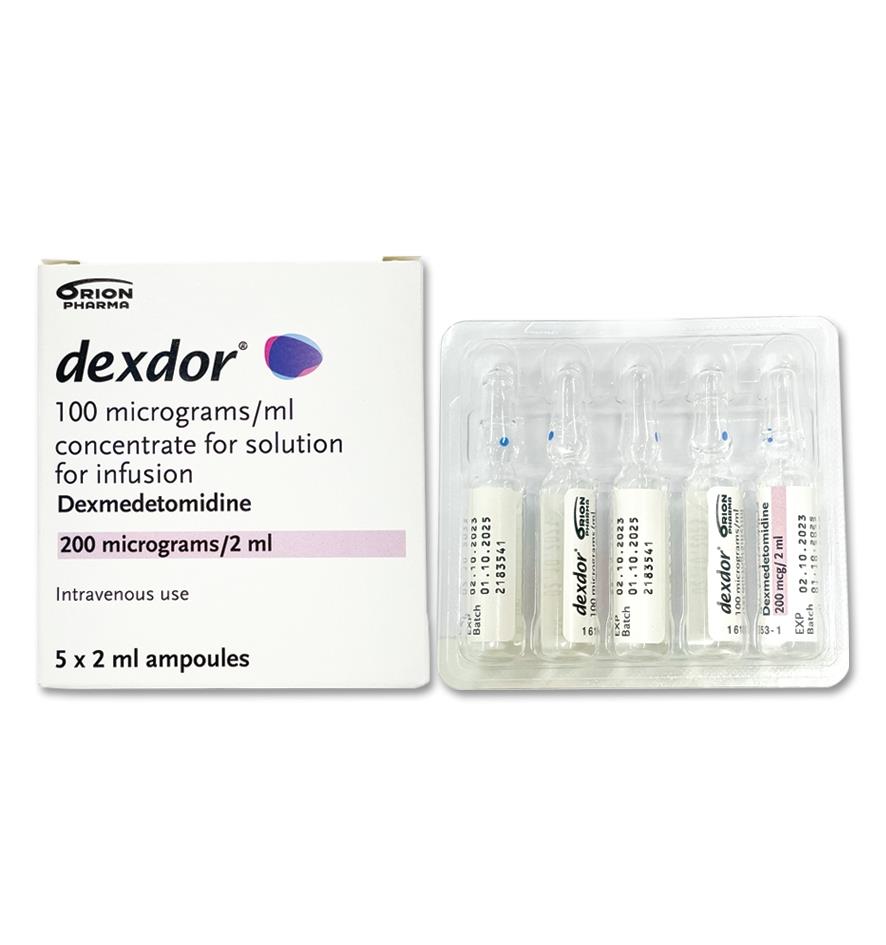
Dexmedetomidine exhibits linear pharmacokinetics in the dosage range of 0.2 to 0.7 mcg/kg/hr when administered by intravenous infusion for up to 24 hours. Table 1 shows the main pharmacokinetic parameters when dexmedetomidine was infused (after appropriate loading doses) at maintenance infusion rates of 0.17 mcg/kg/hr (target plasma concentration of 0.3 ng/mL) for 12 and 24 hours, 0.33 mcg/kg/hr (target plasma concentration of 0.6 ng/mL) for 24 hours, and 0.70 mcg/kg/hr (target plasma concentration of 1.25 ng/mL) for 24 hours. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe loading doses for each of the previously mentioned indicated groups were 0.5, 0.5, 1 and 2.2 mcg/kg, respectively.
Dexmedetomidine pharmacokinetic parameters after dexmedetomidine maintenance doses of 0.2 to 1.4 mcg/kg/hr for >24 hours were similar to the PK parameters after dexmedetomidine maintenance dosing for <24 hours in other studies. The values for clearance (CL), volume of distribution (V), and t½ were 39.4 L/hr, 152 L, and 2.67 hours, respectively.
Distribution: The steady-state volume of distribution (Vss) of dexmedetomidine was approximately 118 liters. Dexmedetomidine protein binding was assessed in the plasma of normal healthy male and female subjects. The average protein binding was 94% and was constant across the different plasma concentrations tested. Protein binding was similar in males and females. The fraction of Dexdor that was bound to plasma proteins was significantly decreased in subjects with hepatic impairment compared to healthy subjects.
The potential for protein binding displacement of dexmedetomidine by fentanyl, ketorolac, theophylline, digoxin and lidocaine was explored in vitro, and negligible changes in the plasma protein binding of Dexdor were observed. The potential for protein binding displacement of phenytoin, warfarin, ibuprofen, propranolol, theophylline and digoxin by Dexdor was explored in vitro and none of these compounds appeared to be significantly displaced by Dexdor.
Metabolism: Dexmedetomidine undergoes almost complete biotransformation with very little unchanged dexmedetomidine excreted in urine and feces. Biotransformation involves both direct glucuronidation as well as cytochrome P450 mediated metabolism. The major metabolic pathways of dexmedetomidine are: direct N-glucuronidation to inactive metabolites; aliphatic hydroxylation (mediated primarily by CYP2A6 with a minor role of CYP1A2, CYP2E1, CYP2D6 and CYP2C19) of dexmedetomidine to generate 3-hydroxydexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3-carboxydexmedetomidine; and N methylation of dexmedetomidine to generate 3-hydroxy N-methyldexmedetomidine, 3-carboxy N-methyl-dexmedetomidine, and dexmedetomidine-N-methyl O glucuronide.
Elimination: The terminal elimination half-life (t½) of dexmedetomidine is approximately 2 hours and clearance is estimated to be approximately 39 L/h. A mass balance study demonstrated that after nine days an average of 95% of the radioactivity, following intravenous administration of radiolabeled dexmedetomidine, was recovered in the urine and 4% in the feces. No unchanged dexmedetomidine was detected in the urine. Approximately 85% of the radioactivity recovered in the urine was excreted within 24 hours after the infusion. Fractionation of the radioactivity excreted in urine demonstrated that products of N-glucuronidation accounted for approximately 34% of the cumulative urinary excretion. In addition, aliphatic hydroxylation of parent drug to form 3-hydroxy-dexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3-carboxylic acid-dexmedetomidine together represented approximately 14% of the dose in urine. N-methylation of dexmedetomidine to form 3 hydroxy N-methyl dexmedetomidine, 3-carboxy N-methyl dexmedetomidine, and N methyl O glucuronide dexmedetomidine accounted for approximately 18% of the dose in urine. The N Methyl metabolite itself was a minor circulating component and was undetected in urine. Approximately 28% of the urinary metabolites have not been identified.
Gender: There was no observed difference in Dexdor pharmacokinetics due to gender.
Geriatrics: The pharmacokinetic profile of Dexdor was not altered by age. There were no differences in the pharmacokinetics of Dexdor in young (18 - 40 years), middle age (41 - 65 years), and elderly (>65 years) subjects.
Hepatic Impairment: In subjects with varying degrees of hepatic impairment (Child-Pugh Class A, B, or C), clearance values for Dexdor were lower than in healthy subjects. The mean clearance values for patients with mild, moderate, and severe hepatic impairment were 74%, 64% and 53% of those observed in the normal healthy subjects, respectively. Mean clearances for free drug were 59%, 51% and 32% of those observed in the normal healthy subjects, respectively.
Although Dexdor is dosed to effect, it may be necessary to consider dose reduction in subjects with hepatic impairment [see Dosage Information under Dosage & Administration and Hepatic Impairment under Precautions].
Renal Impairment: Dexdor pharmacokinetics (Cmax, Tmax, AUC, t½, CL, and Vss) were not significantly different in patients with severe renal impairment (creatinine clearance: <30 mL/min) compared to healthy subjects.
Drug Interactions: In vitro studies: In vitro studies in human liver microsomes demonstrated no evidence of cytochrome P450 mediated drug interactions that are likely to be of clinical relevance.
NONCLINICAL TOXICOLOGY: Carcinogenesis, Mutagenesis, Impairment of Fertility: Animal carcinogenicity studies have not been performed with Dexdor.
Dexmedetomidine was not mutagenic in vitro, in either the bacterial reverse mutation assay (E. coli and Salmonella typhimurium) or the mammalian cell forward mutation assay (mouse lymphoma). Dexmedetomidine was clastogenic in the in vitro human lymphocyte chromosome aberration test with, but not without, rat S9 metabolic activation. In contrast, dexmedetomidine was not clastogenic in the in vitro human lymphocyte chromosome aberration test with or without human S9 metabolic activation. Although dexmedetomidine was clastogenic in an in vivo mouse micronucleus test in NMRI mice, there was no evidence of clastogenicity in CD-1 mice.
Fertility in male or female rats was not affected after daily subcutaneous injections of dexmedetomidine at doses up to 54 mcg/kg (less than the maximum recommended human intravenous dose on a mcg/m2 basis) administered from 10 weeks prior to mating in males and 3 weeks prior to mating and during mating in females.
Animal Pharmacology and/or Toxicology: There were no differences in the adrenocorticotropic hormone (ACTH)-stimulated cortisol response in dogs following a single dose of dexmedetomidine compared to saline control. However, after continuous subcutaneous infusions of Dexdor at 3 mcg/kg/hr and 10 mcg/kg/hr for one week in dogs (exposures estimated to be within the clinical range), the ACTH-stimulated cortisol response was diminished by approximately 27% and 40%, respectively, compared to saline-treated control animals indicating a dose-dependent adrenal suppression.
CLINICAL STUDIES: The safety and efficacy of Dexdor has been evaluated in four randomized, double-blind, placebo-controlled multicenter clinical trials in 1185 adult patients.
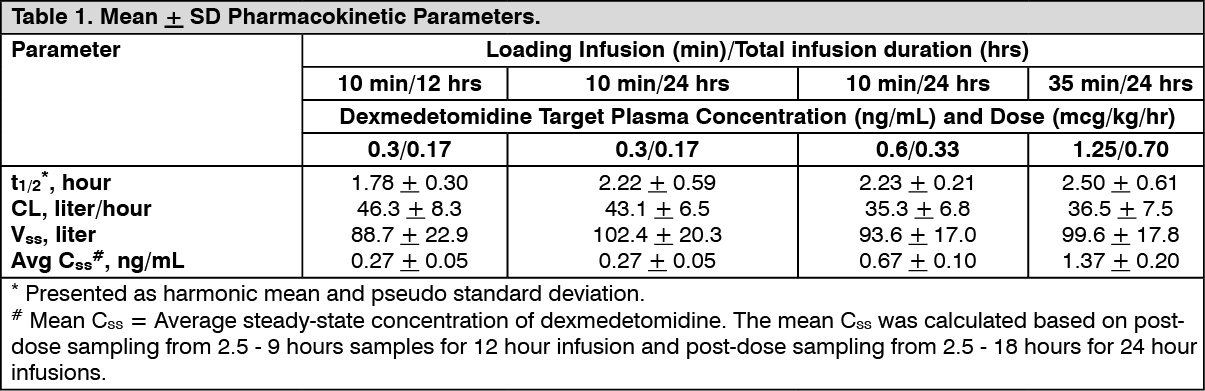
Intensive Care Unit Sedation: Two randomized, double-blind, parallel-group, placebo-controlled multicenter clinical trials included 754 adult patients being treated in a surgical intensive care unit. All patients were initially intubated and received mechanical ventilation. These trials evaluated the sedative properties of dexmedetomidine by comparing the amount of rescue medication (midazolam in one trial and propofol in the second) required to achieve a specified level of sedation (using the standardized Ramsay sedation Scale) between dexmedetomidine and placebo from onset of treatment to extubation or to a total treatment duration of 24 hours. The Ramsay Level of Sedation Scale is displayed in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the first study, 175 adult patients were randomized to receive placebo and 178 to receive dexmedetomidine by intravenous infusion at a dose of 0.4 mcg/kg/hr (with allowed adjustment between 0.2 and 0.7 mcg/kg/hr) following an initial loading infusion of one mcg/kg intravenous over 10 minutes. The study drug infusion rate was adjusted to maintain a Ramsay sedation score of ≥3. Patients were allowed to receive "rescue" midazolam as needed to augment the study drug infusion. In addition, morphine sulfate was administered for pain as needed. The primary outcome measure for this study was the total amount of rescue medication (midazolam) needed to maintain sedation as specified while intubated. Patients randomized to placebo received significantly more midazolam than patients randomized to dexmedetomidine (see Table 3).
A second prospective primary analysis assessed the sedative effects of dexmedetomidine by comparing the percentage of patients who achieved a Ramsay sedation score of ≥3 during intubation without the use of additional rescue medication. A significantly greater percentage of patients in the dexmedetomidine group maintained a Ramsay sedation score of ≥3 without receiving any midazolam rescue compared to the placebo group (see Table 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA prospective secondary analysis assessed the dose of morphine sulfate administered to patients in the dexmedetomidine and placebo groups. On average, dexmedetomidine-treated patients received less morphine sulfate for pain than placebo treated patients (0.47 versus 0.83 mg/h). In addition, 44% (79 of 178 patients) of dexmedetomidine patients received no morphine sulfate for pain versus 19% (33 of 175 patients) in the placebo group.
In a second study, 198 adult patients were randomized to receive placebo and 203 to receive dexmedetomidine by intravenous infusion at a dose of 0.4 mcg/kg/hr (with allowed adjustment between 0.2 and 0.7 mcg/kg/hr) following an initial loading infusion of one mcg/kg intravenous over 10 minutes. The study drug infusion was adjusted to maintain a Ramsay sedation score of ≥3. Patients were allowed to receive "rescue" propofol as needed to augment the study drug infusion. In addition, morphine sulfate was administered as needed for pain. The primary outcome measure for this study was the total amount of rescue medication (propofol) needed to maintain sedation as specified while intubated.
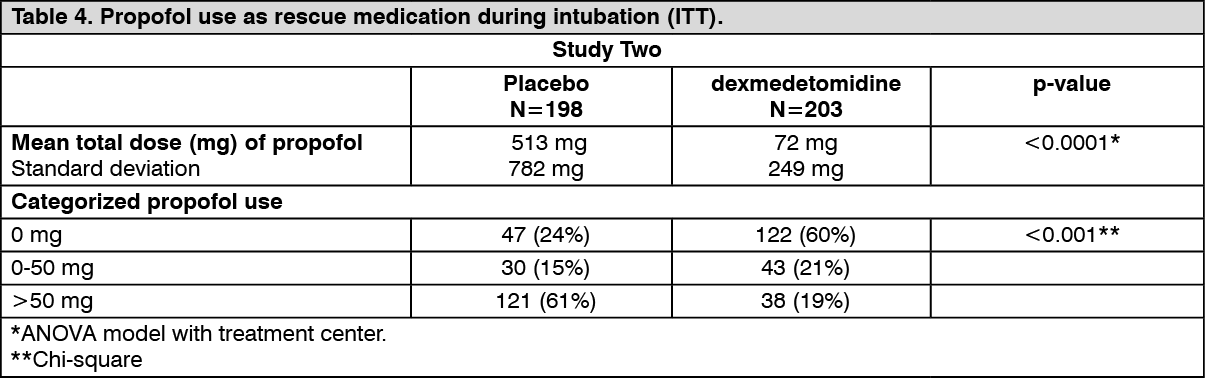
Patients randomized to placebo received significantly more propofol than patients randomized to dexmedetomidine (see Table 4).
A significantly greater percentage of patients in the dexmedetomidine group compared to the placebo group maintained a Ramsay sedation score of ≥3 without receiving any propofol rescue (see Table 4).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA prospective secondary analysis assessed the dose of morphine sulfate administered to patients in the dexmedetomidine and placebo groups. On average, dexmedetomidine-treated patients received less morphine sulfate for pain than placebo treated patients (0.43 versus 0.89 mg/h). In addition, 41% (83 of 203 patients) of dexmedetomidine patients received no morphine sulfate for pain versus 15% (30 of 198 patients) in the placebo group.
In a controlled clinical trial, dexmedetomidine was compared to midazolam for ICU sedation exceeding 24 hours duration. Dexmedetomidine was not shown to be superior to midazolam for the primary efficacy endpoint, the percent of time patients were adequately sedated (81% versus 81%). In addition, administration of dexmedetomidine for longer than 24 hours was associated with tolerance, tachyphylaxis, and a dose-related increase in adverse events [see Clinical Studies Experience under Adverse Reactions].
In two randomized, double-blind, parallel-group, comparator-controlled multicenter clinical trials 1000 adult patients were randomized to either continue the current sedative agent or be switched to dexmedetomidine. The two trials evaluated dexmedetomidine compared to propofol or midazolam for sedation in intubated and mechanically ventilated ICU patients, who required sedation over 24 hours. The first hierarchical primary objective was to demonstrate that dexmedetomidine is at least as effective as sedation with midazolam or propofol in maintaining a target depth of sedation. The dose range of dexmedetomidine was 0.2-1.4 mcg/kg/h, and the length of sedation was up to 14 days. The target sedation range was light to moderate, corresponding to Richmond Agitation Sedation Scale 0 to -3. The Richmond Agitation Sedation Scale is displayed in Table 5. (See Table 5.)
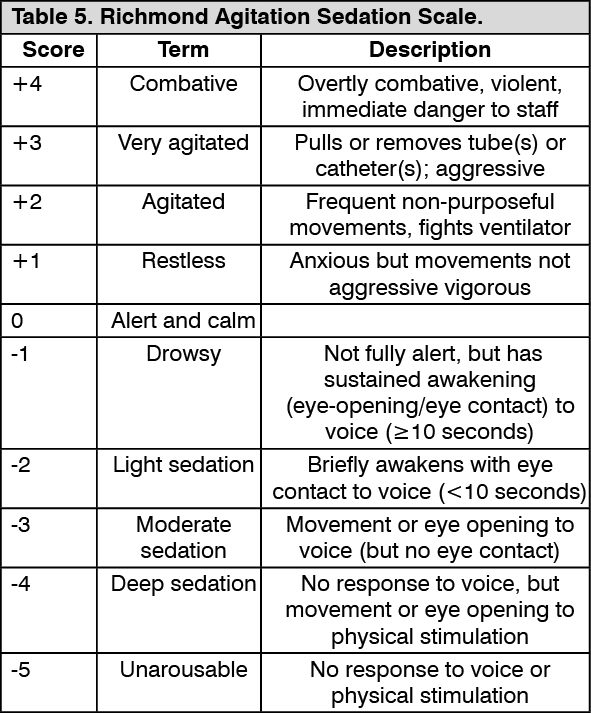 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the first study, 249 adult patients were randomized to receive propofol and 251 to receive dexmedetomidine by intravenous infusion. Loading doses were not used, the starting infusion dose for the first hour was between 0.2 and 0.7 mcg/kg/h then titrated between 0.2 and 1.4 mcg/kg/h to maintain RASS scores between 0 and -3. The first-line rescue medication for inadequate sedation was midazolam, for analgesia fentanyl was used. The co-primary efficacy end points were 1. the proportion of time in target sedation range without use of rescue therapy, 2. the duration of mechanical ventilation. Secondary efficacy outcomes were length of ICU stay and nurses' assessment of arousal, ability to cooperate with care, and ability to communicate pain using visual analogue scales (VAS). The mean percentage of time at target sedation level without use of rescue medication was 64.6% and 64.7% for dexmedetomidine and propofol, respectively, dexmedetomidine was demonstrated to be as effective as propofol. The median duration of mechanical ventilation was 96.5 and 117.5 hours in the dexmedetomidine and propofol groups, respectively (p=0.240). Subjects on dexmedetomidine were significantly (p < 0.001) more arousable, cooperative and better able to communicate whether they had pain than those on propofol. The median length of ICU stay was 6.8 and 7.7 days for dexmedetomidine and propofol, respectively (p = 0.535). 72.5% of subjects in the dexmedetomidine than 64.4% of subjects in the propofol group used the first-line (i.e. midazolam) rescue treatment for inadequate sedation (p = 0.054). The mean total dose (32.9 vs. 22.8 mg, p=0.024) of midazolam was higher for dexmedetomidine.
In the second study, 251 adult patients were randomized to receive midazolam and 249 to receive dexmedetomidine by intravenous infusion. Loading doses were not used, the starting infusion dose for the first hour was between 0.2 and 0.7 mcg/kg/h then titrated between 0.2 and 1.4 mcg/kg/h to maintain RASS scores between 0 and -3. The first-line rescue medication for inadequate sedation was propofol, for analgesia fentanyl was used. The co-primary efficacy end points and the secondary efficacy outcomes were identical than in the first study. The mean percentage of time at target sedation level without use of rescue medication was 60.7% and 56.6% for dexmedetomidine and midazolam, respectively, dexmedetomidine was demonstrated to be as effective as midazolam. The median duration of mechanical ventilation was 123.0 and 164.0 hours in the dexmedetomidine and midazolam groups, respectively (p = 0.033 Gehan-Wilcoxon; p = 0.265, Cox's proportional-hazards regression). Subjects on dexmedetomidine were significantly (p < 0.001) more arousable, cooperative and better able to communicate whether they had pain than those on midazolam. The median length of ICU stay was 8.8 and 10.1 days for dexmedetomidine and midazolam, respectively (p = 0.269). A similar percentage of dexmedetomidine (43.8%) and midazolam subjects (45.4%) used the first-line rescue treatment (propofol) for inadequate sedation. Similarly, no difference between groups in the mean total dose of propofol used (360 vs. 299 mg, p = 0.317) was observed.
Procedural Sedation: The safety and efficacy of dexmedetomidine for sedation of non-intubated patients prior to and/or during surgical and other procedures was evaluated in two randomized, double-blind, placebo-controlled multicenter clinical trials. Study 1 evaluated the sedative properties of dexmedetomidine in patients having a variety of elective surgeries/procedures performed under monitored anesthesia care. Study 2 evaluated dexmedetomidine in patients undergoing awake fiberoptic intubation prior to a surgical or diagnostic procedure.
In Study 1, the sedative properties of dexmedetomidine were evaluated by comparing the percent of patients not requiring rescue midazolam to achieve a specified level of sedation using the standardized Observer's Assessment of Alertness/Sedation Scale (see Table 6).
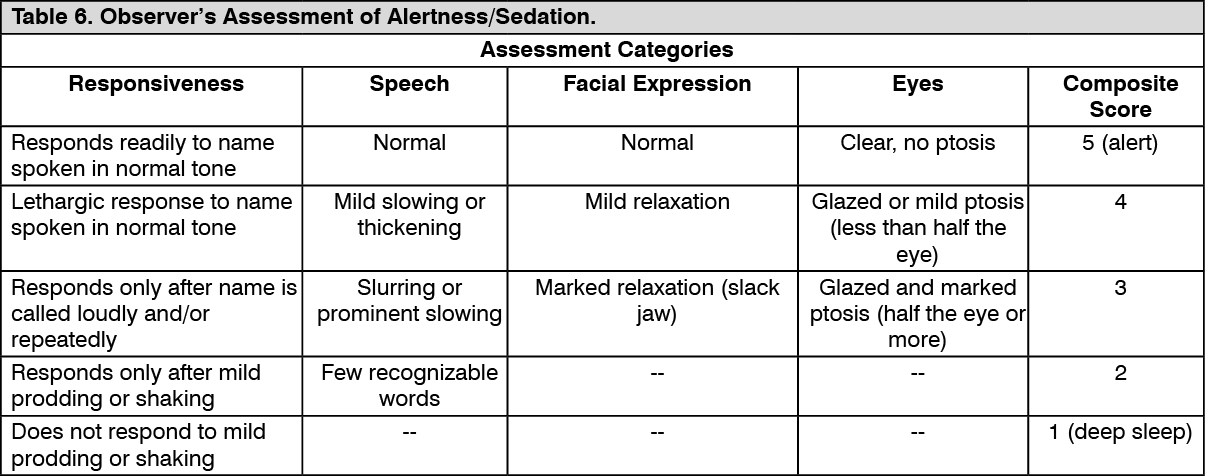 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients were randomized to receive a loading infusion of either dexmedetomidine 1 mcg/kg, dexmedetomidine 0.5 mcg/kg, or placebo (normal saline) given over 10 minutes and followed by a maintenance infusion started at 0.6 mcg/kg/hr. The maintenance infusion of study drug could be titrated from 0.2 mcg/kg/hr to 1 mcg/kg/hr to achieve the targeted sedation score (Observer's Assessment of Alertness/Sedation Scale ≤4). Patients were allowed to receive rescue midazolam as needed to achieve and/or maintain an Observer's Assessment of Alertness/Sedation Scale ≤4. After achieving the desired level of sedation, a local or regional anesthetic block was performed. Demographic characteristics were similar between the dexmedetomidine and comparator groups. Efficacy results showed that dexmedetomidine was more effective than the comparator group when used to sedate non-intubated patients requiring monitored anesthesia care during surgical and other procedures (see Table 7).
In Study 2, the sedative properties of dexmedetomidine were evaluated by comparing the percent of patients requiring rescue midazolam to achieve or maintain a specified level of sedation using the Ramsay Sedation Scale score ≥2 (see Table 2). Patients were randomized to receive a loading infusion of dexmedetomidine 1 mcg/kg or placebo (normal saline) given over 10 minutes and followed by a fixed maintenance infusion of 0.7 mcg/kg/hr. After achieving the desired level of sedation, topicalization of the airway occurred. Patients were allowed to receive rescue midazolam as needed to achieve and/or maintain a Ramsay Sedation Scale ≥2. Demographic characteristics were similar between the dexmedetomidine and comparator groups. For efficacy results see Table 7.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image