Pharmacotherapeutic group: Antivirals for systemic use, protease inhibitors.
ATC code: Not yet assigned.
Namibia Pharmacological Classification: 20.2.8 - Antiviral agents.
Pharmacology: Pharmacodynamics: Mechanism of action: Darunavir is an inhibitor of the dimerisation and of the catalytic activity of the HIV-1 protease (KD of 4.5 x 10-12M). It selectively inhibits the cleavage of HIV encoded Gag-Pol polyproteins in virus infected cells, thereby preventing the formation of mature infectious virus particles.
Antiviral activity in vitro: Darunavir exhibits activity against laboratory strains and clinical isolates of HIV-1 and laboratory strains of HIV-2 in acutely infected T-cell lines, human peripheral blood mononuclear cells and human monocytes/macrophages with median EC
50 values ranging from 1.2 to 8.5 nM (0.7 to 5.0 ng/ml). Darunavir demonstrates antiviral activity
in vitro against a broad panel of HIV-1 group M (A, B, C, D, E, F, G) and group O primary isolates with EC
50 values ranging from <0.1 to 4.3 nM.
These EC
50 values are well below the 50% cellular toxicity concentration range of 87 μM to >100 μM.
Resistance: In vitro selection of darunavir-resistant virus from wild type HIV-1 was lengthy (>3 years). The selected viruses were unable to grow in the presence of darunavir concentrations above 400 nM. Viruses selected in these conditions and showing decreased susceptibility to darunavir (range: 23-50-fold) harboured 2 to 4 amino acid substitutions in the protease gene. The decreased susceptibility to darunavir of the emerging viruses in the selection experiment could not be explained by the emergence of these protease mutations.
The clinical trial data from ART-experienced patients (TITAN trial and the pooled analysis of the POWER 1, 2 and 3 and DUET 1 and 2 trials) showed that virologic response to Darunavir co-administered with low dose ritonavir was decreased when 3 or more darunavir RAMs (V11I, V32I, L33F, I47V, I50V, I54L or M, T74P, L76V, I84V and L89V) were present at baseline or when these mutations developed during treatment.
Increasing baseline darunavir fold change in EC50
(FC) was associated with decreasing Virologic response. A lower and upper clinical cut-off of 10 and 40 were identified. Isolates with baseline FC ≤10 are susceptible; isolates with FC >10 to 40 have decreased susceptibility; isolates with FC >40 are resistant (see Clinical results as follows).
Viruses isolated from patients on Darunavir/ritonavir 600/100 mg twice daily experiencing Virologic failure by rebound that were susceptible to tipranavir at baseline remained susceptible to Tipranavir after treatment in the vast majority of cases.
The lowest rates of developing resistant HIV virus are observed in ART-naïve patients who are treated for the first time with darunavir in combination with other ART.
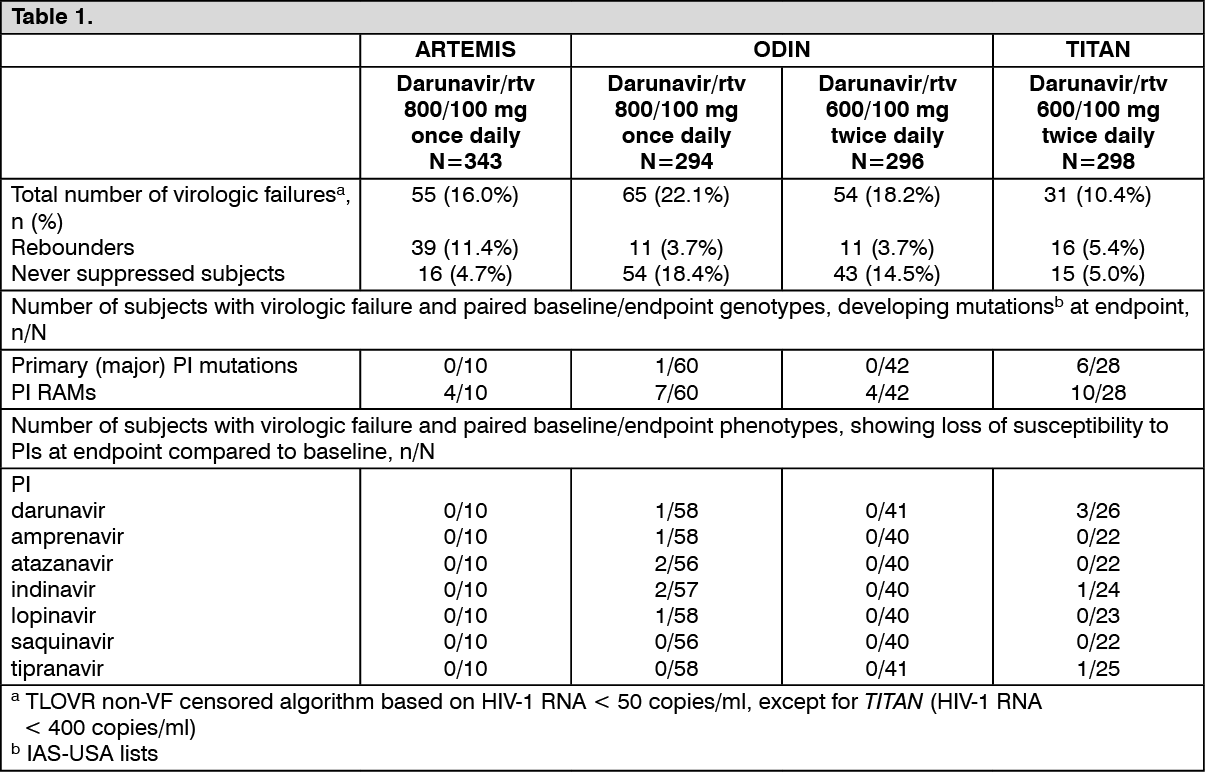
The table as follows shows the development of HIV-1 protease mutations and loss of susceptibility to PIs in virologic failures at endpoint in the ARTEMIS, ODIN and TITAN trials. (See Table 1.)
 Click on icon to see table/diagram/image
Cross-resistance:
Click on icon to see table/diagram/image
Cross-resistance: Darunavir FC was less than 10 for 90% of 3,309 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and/or tipranavir showing that viruses resistant to most PIs remain susceptible to darunavir.
In the virologic failures of the ARTEMIS trial no cross-resistance with other PIs was observed.
Clinical results: Efficacy of Darunavir 800 mg once daily co-administered with 100 mg ritonavir once daily in ART-naïve patients: The evidence of efficacy of Darunavir/ritonavir 800/100 mg once daily is based on the analyses of 192 week data from the randomised, controlled, open-label Phase III trial ARTEMIS in antiretroviral treatment-naïve HIV-1 infected patients comparing Darunavir/ritonavir 800/100 mg once daily with lopinavir/ritonavir 800/200 mg per day (given as a twice-daily or as a once-daily regimen). Both arms used a fixed background regimen consisting of tenofovir disoproxil fumarate 300 mg once daily and emtricitabine 200 mg once daily.
The table as follows shows the efficacy data of the 48 week and 96 week analyses from the ARTEMIS
trial: See Table 2.
Click on icon to see table/diagram/image
Non-inferiority in virologic response to the Darunavir/ritonavir treatment, defined as the percentage of patients with plasma HIV-1 RNA level <50 copies/ml, was demonstrated (at the pre-defined 12% non-inferiority margin) for both Intent-To-Treat (ITT) and On Protocol (OP) populations in the 48 week analysis. These results were confirmed in the analyses of data at 96 weeks of treatment in the ARTEMIS trial. These results were sustained up to 192 weeks of treatment in the ARTEMIS trial.
Efficacy of Darunavir 800 mg once daily co-administered with 100 mg ritonavir once daily in ART-experienced patients: ODIN is a Phase III, randomised, open-label trial comparing Darunavir/ritonavir 800/100 mg once daily versus Darunavir/ritonavir 600/100 mg twice daily in ART-experienced HIV-1 infected patients with screening genotype resistance testing showing no darunavir RAMs (i.e. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V) and a screening HIV-1 RNA >1,000 copies/ml. Efficacy analysis is based on 48 weeks of treatment (see table as follows). Both arms used an optimised background regimen (OBR) of ≥2 NRTIs. (See Table 3.)
Click on icon to see table/diagram/image
At 48 weeks, virologic response, defined as the percentage of patients with plasma HIV-1 RNA level <50 copies/ml, with Darunavir/ritonavir 800/100 mg once daily treatment was demonstrated to be non-inferior (at the pre-defined 12% non-inferiority margin) compared to Darunavir/ritonavir 600/100 mg twice daily for both ITT and OP populations.
Darunavir/ritonavir 800/100 mg once daily in ART-experienced patients should not be used in patients with one or more darunavir resistance associated mutations (DRV-RAMs) or HIV-1 RNA ≥100,000 copies/ml or CD4+ cell count <100 cells x 10
6/l (see Dosage & Administration and Precautions). Limited data is available in patients with HIV-1 clades other than B.
Paediatric patients: ART-naïve paediatric patients from the age of 12 years to <18 years, and weighing at least 40 kg: DIONE is an open-label, Phase II trial evaluating the pharmacokinetics, safety, tolerability, and efficacy of Darunavir with low dose ritonavir in 12 ART-naïve HIV-1 infected paediatric patients aged 12 to less than 18 years and weighing at least 40 kg. These patients received Darunavir/ritonavir 800/100 mg once daily in combination with other antiretroviral agents. Virologic response was defined as a decrease in plasma HIV-1 RNA viral load of at least 1.0 log10 versus baseline. (See Table 4.)
Click on icon to see table/diagram/image
Pregnancy and postpartum: Darunavir/ritonavir (600/100 mg twice daily or 800/100 mg once daily) in combination with a background regimen was evaluated in a clinical trial of 34 pregnant women (17 in each arm) during the second and third trimesters, and postpartum. Virologic response was preserved throughout the study period in both arms. No mother to child transmission occurred in the infants born to the 29 subjects who stayed on the antiretroviral treatment through delivery. There were no new clinically relevant safety findings compared with the known safety profile of darunavir/ritonavir in HIV-1 infected adults (see Dosage & Administration, Precautions and Pharmacokinetics as follows).
Pharmacokinetics: Darunavir: The pharmacokinetic properties of darunavir, co-administered with ritonavir, have been evaluated in healthy adult volunteers and in HIV-1 infected patients. Exposure to darunavir was higher in HIV-1 infected patients than in healthy subjects. The increased exposure to darunavir in HIV-1 infected patients compared to healthy subjects may be explained by the higher concentrations of α
1-acid glycoprotein (AAG) in HIV-1 infected patients, resulting in higher darunavir binding to plasma AAG and, therefore, higher plasma concentrations.
Darunavir is primarily metabolised by CYP3A. Ritonavir inhibit CYP3A, thereby increasing the plasma concentrations of darunavir considerably.
Absorption: Darunavir was rapidly absorbed following oral administration. Maximum plasma concentration of darunavir in the presence of low dose ritonavir is generally achieved within 2.5-4.0 hours.
The absolute oral bioavailability of a single 600 mg dose of darunavir alone was approximately 37% and increased to approximately 82% in the presence of 100 mg twice daily ritonavir. The overall pharmacokinetic enhancement effect by ritonavir was an approximate 14-fold increase in the systemic exposure of darunavir when a single dose of 600 mg darunavir was given orally in combination with ritonavir at 100 mg twice daily (see Precautions).
When administered without food, the relative bioavailability of darunavir in the presence of low dose ritonavir is lower as compared to intake with food. Therefore, Darunavir tablets should be taken with ritonavir and with food. The type of food does not affect exposure to darunavir.
Distribution: Darunavir is approximately 95% bound to plasma protein. Darunavir binds primarily to plasma α
1-acid glycoprotein.
Following intravenous administration, the volume of distribution of darunavir alone was 88.1 ± 59.0 l (Mean ± SD) and increased to 131 ± 49.9 l (Mean ± SD) in the presence of 100 mg twice-daily ritonavir.
Biotransformation:
In vitro experiments with human liver microsomes (HLMs) indicate that darunavir primarily undergoes oxidative metabolism. Darunavir is extensively metabolised by the hepatic CYP system and almost exclusively by isozyme CYP3A4. A
14C-darunavir trial in healthy volunteers showed that a majority of the radioactivity in plasma after a single 400/100 mg darunavir with ritonavir dose was due to the parent active substance. At least 3 oxidative metabolites of darunavir have been identified in humans; all showed activity that was at least 10-fold less than the activity of darunavir against wild type HIV.
Elimination: After a 400/100 mg
14C-darunavir with ritonavir dose, approximately 79.5% and 13.9% of the administered dose of
14C-darunavir could be retrieved in faeces and urine, respectively. Unchanged darunavir accounted for approximately 41.2% and 7.7% of the administered dose in faeces and urine, respectively. The terminal elimination half-life of darunavir was approximately 15 hours when combined with ritonavir.
The intravenous clearance of darunavir alone (150 mg) and in the presence of low dose ritonavir was 32.8 l/h and 5.9 l/h, respectively.
Special populations: Paediatric population: The pharmacokinetics of darunavir in combination with ritonavir taken twice daily in 74 treatment-experienced paediatric patients, aged 6 to 17 years and weighing at least 20 kg, showed that the administered weight-based doses of Darunavir/ritonavir resulted in darunavir exposure comparable to that in adults receiving Darunavir/ritonavir 600/100 mg twice daily (see Dosage & Administration).
The pharmacokinetics of darunavir in combination with ritonavir taken twice daily in 14 treatment-experienced paediatric patients, aged 3 to <6 years and weighing at least 15 kg to <20 kg, showed that weight-based dosages resulted in darunavir exposure that was comparable to that achieved in adults receiving Darunavir/ritonavir 600/100 mg twice daily (see Dosage & Administration).
The pharmacokinetics of darunavir in combination with ritonavir taken once daily in 12 ART-naïve paediatric patients, aged 12 to <18 years and weighing at least 40 kg, showed that Darunavir/ritonavir 800/100 mg once daily results in darunavir exposure that was comparable to that achieved in adults receiving Darunavir/ritonavir 800/100 mg once daily. Therefore the same once daily dosage may be used in treatment-experienced adolescents aged 12 to <18 years and weighing at least 40 kg without darunavir resistance associated mutations (DRV-RAMs)* and who have plasma HIV-1 RNA <100,000 copies/ml and CD4+ cell count ≥100 cells x 10
6/l (see Dosage & Administration).
* DRV-RAMs: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V and L89V.
The pharmacokinetics of darunavir in combination with ritonavir taken once daily in 10 treatment-experienced paediatric patients, aged 3 to <6 years and weighing at least 14 kg to <20 kg, showed that weight-based dosages resulted in darunavir exposure that was comparable to that achieved in adults receiving Darunavir/ritonavir 800/100 mg once daily (see Dosage & Administration). In addition, pharmacokinetic modeling and simulation of darunavir exposures in paediatric patients across the ages of 3 to <18 years confirmed the darunavir exposures as observed in the clinical studies and allowed the identification of weight-based Darunavir/ritonavir once daily dosing regimens for paediatric patients weighing at least 15 kg that are either ART-naïve or treatment-experienced paediatric patients without DRV-RAMs* and who have plasma HIV-1 RNA <100,000 copies/ml and CD4+ cell count ≥100 cells x 10
6/l (see Dosage & Administration).
* DRV-RAMs: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V and L89V.
Elderly: Population pharmacokinetic analysis in HIV infected patients showed that darunavir pharmacokinetics are not considerably different in the age range (18 to 75 years) evaluated in HIV infected patients (n=12, age 65) (see Precautions). However, only limited data were available in patients above the age of 65 year.
Gender: Population pharmacokinetic analysis showed a slightly higher darunavir exposure (16.8%) in HIV infected females compared to males. This difference is not clinically relevant.
Renal impairment: Results from a mass balance study with
14C-darunavir with ritonavir showed that approximately 7.7% of the administered dose of darunavir is excreted in the urine unchanged.
Although darunavir has not been studied in patients with renal impairment, population pharmacokinetic analysis showed that the pharmacokinetics of darunavir were not significantly affected in HIV infected patients with moderate renal impairment (CrCl between 30-60 ml/min, n=20) (see Dosage & Administration and Precautions).
Hepatic impairment: Darunavir is primarily metabolised and eliminated by the liver. In a multiple dose study with Darunavir co-administered with ritonavir (600/100 mg) twice daily, it was demonstrated that the total plasma concentrations of darunavir in subjects with mild (Child-Pugh Class A, n=8) and moderate (Child-Pugh Class B, n=8) hepatic impairment were comparable with those in healthy subjects. However, unbound darunavir concentrations were approximately 55% (Child-Pugh Class A) and 100% (Child-Pugh Class B) higher, respectively. The clinical relevance of this increase is unknown therefore, Darunavir should be used with caution. The effect of severe hepatic impairment on the pharmacokinetics of darunavir has not been studied (see Dosage & Administration, Contraindications and Precautions).
Pregnancy and postpartum: The exposure to total darunavir and ritonavir after intake of darunavir/ritonavir 600/100 mg twice daily and darunavir/ritonavir 800/100 mg once daily as part of an antiretroviral regimen was generally lower during pregnancy compared with postpartum. However, for unbound (i.e. active) darunavir, the pharmacokinetic parameters were less reduced during pregnancy compared to postpartum, due to an increase in the unbound fraction of darunavir during pregnancy compared to postpartum. (See Tables 5 and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In women receiving darunavir/ritonavir 600/100 mg twice daily during the second trimester of pregnancy, mean intra-individual values for total darunavir C
max, AUC
12h and C
min were 28%, 24% and 17% lower, respectively, as compared with postpartum; during the third trimester of pregnancy, total darunavir C
max, AUC
12h and C
min values were 19%, 17% lower and 2% higher, respectively, as compared with postpartum.
In women receiving darunavir/ritonavir 800/100 mg once daily during the second trimester of pregnancy, mean intra-individual values for total darunavir C
max, AUC
12h and C
min were 34%, 34% and 32% lower, respectively, as compared with postpartum; during the third trimester of pregnancy, total darunavir C
max, AUC
12h and C
min values were 31%, 35% and 50% lower, respectively, as compared with postpartum.
Ritonavir: Absorption: There is no parenteral formulation of ritonavir, therefore the extent of absorption and absolute bioavailability have not been determined. In healthy subjects (n=44) under fed conditions, using a single dose of two Ritonavir Tablets 100 mg, the following results were obtained (arithmetic mean +/- SD): T
max (h) 5.2+/-1.7, C
max (ng/ml) 3089 +/- 1105, AUC
0-t (ng.h/ml) 22019 +/- 7915, AUC
0-inf (ng.h/ml) 22816 +/- 7993.
Effects of food on oral absorption: Food slightly decreases the bioavailability of the Ritonavir. Administration of a single 100 mg dose of Ritonavir with a moderate fat meal (857 kcal, 31% calories from fat) or a high fat meal (907 kcal, 52% calories from fat) was associated with a mean decrease of 20-23% in ritonavir AUC and C
max.
Distribution:
The apparent volume of distribution (V
B/F) of ritonavir is approximately 20-40 l after a single 600 mg dose. The protein binding of ritonavir in human plasma is approximately 98-99% and is constant over the concentration range of 1.0-100 μg/ml. Ritonavir binds to both human alpha 1-acid glycoprotein (AAG) and human serum albumin (HSA) with comparable affinities.
Tissue distribution studies with
14C-labelled ritonavir in rats showed the liver, adrenals, pancreas, kidneys and thyroid to have the highest concentrations of ritonavir. Tissue to plasma ratios of approximately 1 measured in rat lymph nodes suggests that ritonavir distributes into lymphatic tissues. Ritonavir penetrates minimally into the brain.
Metabolism: Ritonavir was noted to be extensively metabolised by the hepatic cytochrome P450 system, primarily by the CYP3A isozyme family and to a lesser extent by the CYP2D6 isoform. Animal studies as well as
in vitro experiments with human hepatic microsomes indicated that ritonavir primarily underwent oxidative metabolism. Four ritonavir metabolites have been identified in man. The isopropylthiazole oxidation metabolite (M-2) is the major metabolite and has antiviral activity similar to that of parent compound. However, the AUC of the M-2 metabolite was approximately 3% of the AUC of parent compound.
Low doses of Ritonavir have shown profound effects on the pharmacokinetics of other protease inhibitors (and other products metabolised by CYP3A4) and other protease inhibitors may influence the pharmacokinetics of Ritonavir (see Interactions).
Elimination: Human studies with radiolabelled ritonavir demonstrated that the elimination of ritonavir was primarily via the hepatobiliary system; approximately 86% of radiolabel was recovered from stool, part of which is expected to be unabsorbed ritonavir. In these studies renal elimination was not found to be a major route of elimination of ritonavir. This was consistent with the observations in animal studies.
Toxicology: Preclinical safety data: Darunavir: Animal toxicology studies have been conducted at exposures up to clinical exposure levels with darunavir alone, in mice, rats and dogs and in combination with ritonavir in rats and dogs.
In repeated-dose toxicology studies in mice, rats and dogs, there were only limited effects of treatment with darunavir. In rodents the target organs identified were the haematopoietic system, the blood coagulation system, liver and thyroid. A variable but limited decrease in red blood cell-related parameters was observed, together with increases in activated partial thromboplastin time.
Changes were observed in liver (hepatocyte hypertrophy, vacuolation, increased liver enzymes) and thyroid (follicular hypertrophy). In the rat, the combination of darunavir with ritonavir lead to a small increase in effect on RBC parameters, liver and thyroid and increased incidence of islet fibrosis in the pancreas (in male rats only) compared to treatment with darunavir alone. In the dog, no major toxicity findings or target organs were identified up to exposures equivalent to clinical exposure at the recommended dose.
In a study conducted in rats, the number of corpora lutea and implantations were decreased in the presence of maternal toxicity. Otherwise, there were no effects on mating or fertility with darunavir treatment up to 1,000 mg/kg/day and exposure levels below (AUC-0.5 fold) of that in human at the clinically recommended dose. Up to same dose levels, there was no teratogenicity with darunavir in rats and rabbits when treated alone nor in mice when treated in combination with ritonavir. The exposure levels were lower than those with the recommended clinical dose in humans. In a pre- and postnatal development assessment in rats, darunavir with and without ritonavir, caused a transient reduction in body weight gain of the offspring pre-weaning and there was a slight delay in the opening of eyes and ears. Darunavir in combination with ritonavir caused a reduction in the number of pups that exhibited the startle response on day 15 of lactation and a reduced pup survival during lactation. These effects may be secondary to pup exposure to the active substance via the milk and/or maternal toxicity. No post weaning functions were affected with darunavir alone or in combination with ritonavir. In juvenile rats receiving darunavir up to days 23-26, increased mortality was observed with convulsions in some animals. Exposure in plasma, liver and brain was considerably higher than in adult rats after comparable doses in mg/kg between days 5 and 11 of age. After day 23 of life, the exposure was comparable to that in adult rats. The increased exposure was likely at least partly due to immaturity of the drug-metabolising enzymes in juvenile animals. No treatment related mortalities were noted in juvenile rats dosed at 1,000 mg/kg darunavir (single dose) on day 26 of age or at 500 mg/kg (repeated dose) from day 23 to 50 of age, and the exposures and toxicity profile were comparable to those observed in adult rats.
Due to uncertainties regarding the rate of development of the human blood brain barrier and liver enzymes, Darunavir with low dose ritonavir should not be used in paediatric patients below 3 years of age.
Darunavir was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. Daily doses of 150, 450 and 1,000 mg/kg were administered to mice and doses of 50, 150 and 500 mg/kg were administered to rats. Dose-related increases in the incidences of hepatocellular adenomas and carcinomas were observed in males and females of both species. Thyroid follicular cell adenomas were noted in male rats. Administration of darunavir did not cause a statistically significant increase in the incidence of any other benign or malignant neoplasm in mice or rats. The observed hepatocellular and thyroid tumours in rodents are considered to be of limited relevance to humans. Repeated administration of darunavir to rats caused hepatic microsomal enzyme induction and increased thyroid hormone elimination, which predispose rats, but not humans, to thyroid neoplasms. At the highest tested doses, the systemic exposures (based on AUC) to darunavir were between 0.4- and 0.7-fold (mice) and 0.7- and 1-fold (rats), relative to those observed in humans at the recommended therapeutic doses.
After 2 years administration of darunavir at exposures at or below the human exposure, kidney changes were observed in mice (nephrosis) and rats (chronic progressive nephropathy).
Darunavir was not mutagenic or genotoxic in a battery of
in vitro and
in vivo assays including bacterial reverse mutation (Ames), chromosomal aberration in human lymphocytes and
in vivo micronucleus test in mice.
Ritonavir: Repeated dose toxicity studies in animals identified major target organs as the liver, retina, thyroid gland and kidney. Hepatic changes involved hepatocellular, biliary and phagocytic elements and were accompanied by increases in hepatic enzymes. Hyperplasia of the retinal pigment epithelium (RPE) and retinal degeneration have been seen in all of the rodent studies conducted with ritonavir, but have not been seen in dogs. Ultrastructural evidence suggests that these retinal changes may be secondary to phospholipidosis. However, clinical trials revealed no evidence of medicinal product-induced ocular changes in humans. All thyroid changes were reversible upon discontinuation of ritonavir. Clinical investigation in humans has revealed no clinically significant alteration in thyroid function tests. Renal changes including tubular degeneration, chronic inflammation and proteinurea were noted in rats and are felt to be attributable to species-specific spontaneous disease. Furthermore, no clinically significant renal abnormalities were noted in clinical trials.
Developmental toxicity observed in rats (embryolethality, decreased foetal body weight and ossification delays and visceral changes, including delayed testicular descent) occurred mainly at a maternally toxic dosage. Developmental toxicity in rabbits (embryolethality, decreased litter size and decreased foetal weights) occurred at a maternally toxic dosage.
Ritonavir was not found to be mutagenic or clastogenic in a battery of
in vitro and
in vivo assays including the Ames bacterial reverse mutation assay using
S. typhimurium and
E. coli, the mouse lymphoma assay, the mouse micronucleus test and chromosomal aberration assays in human lymphocytes.
Long term carcinogenicity studies of ritonavir in mice and rats revealed tumourigenic potential specific for these species, but are regarded as of no relevance for humans.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image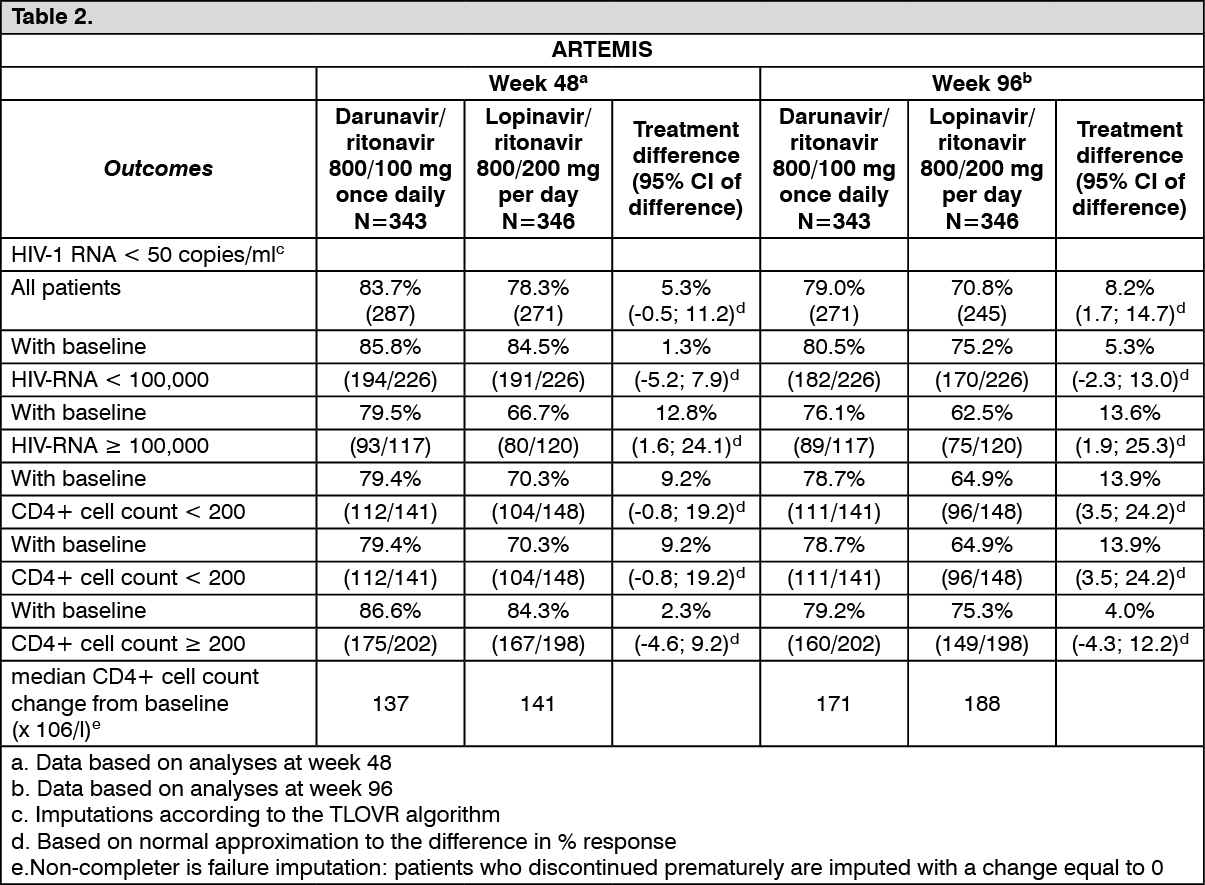 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image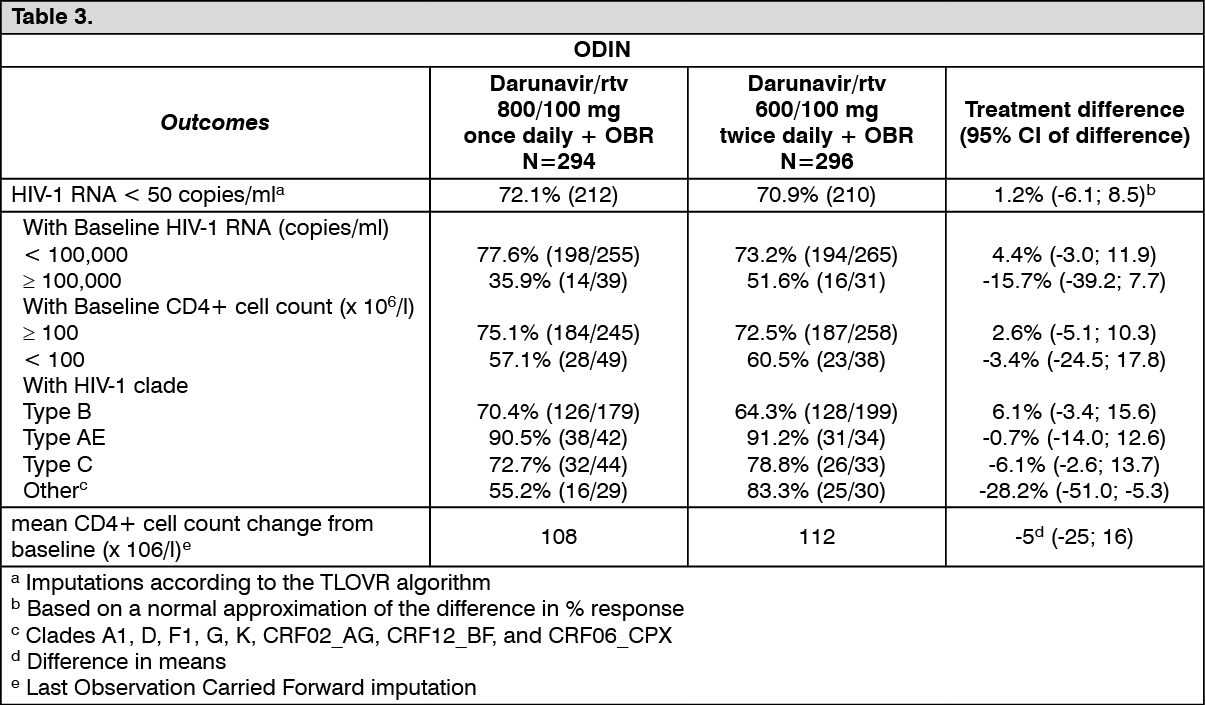 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image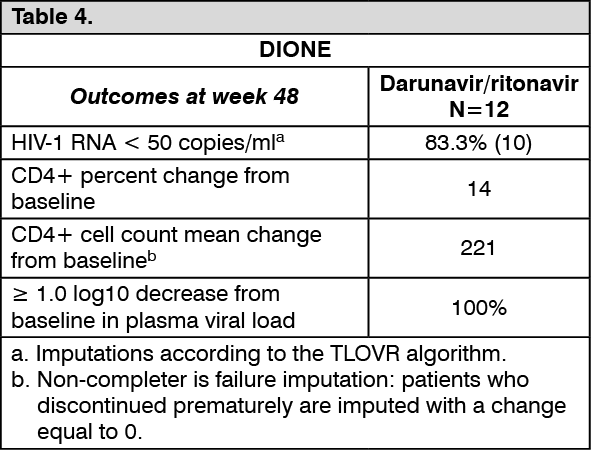 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image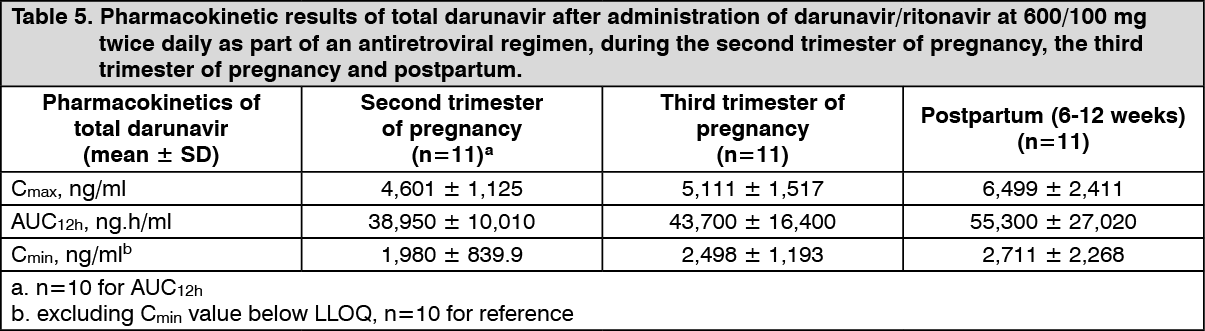 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image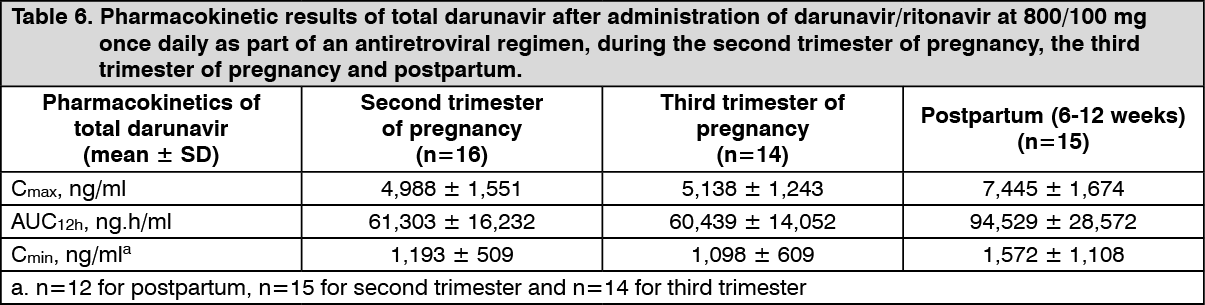 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image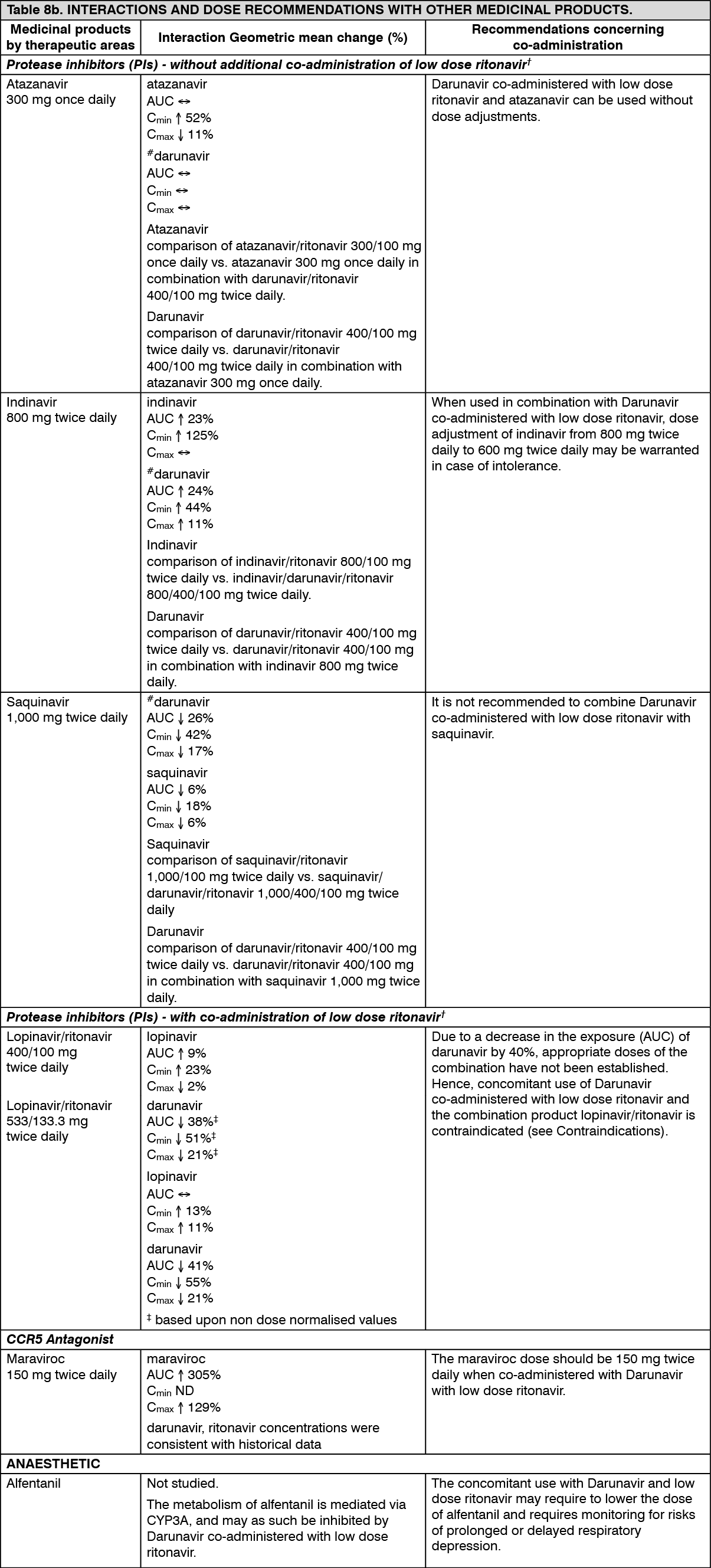 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image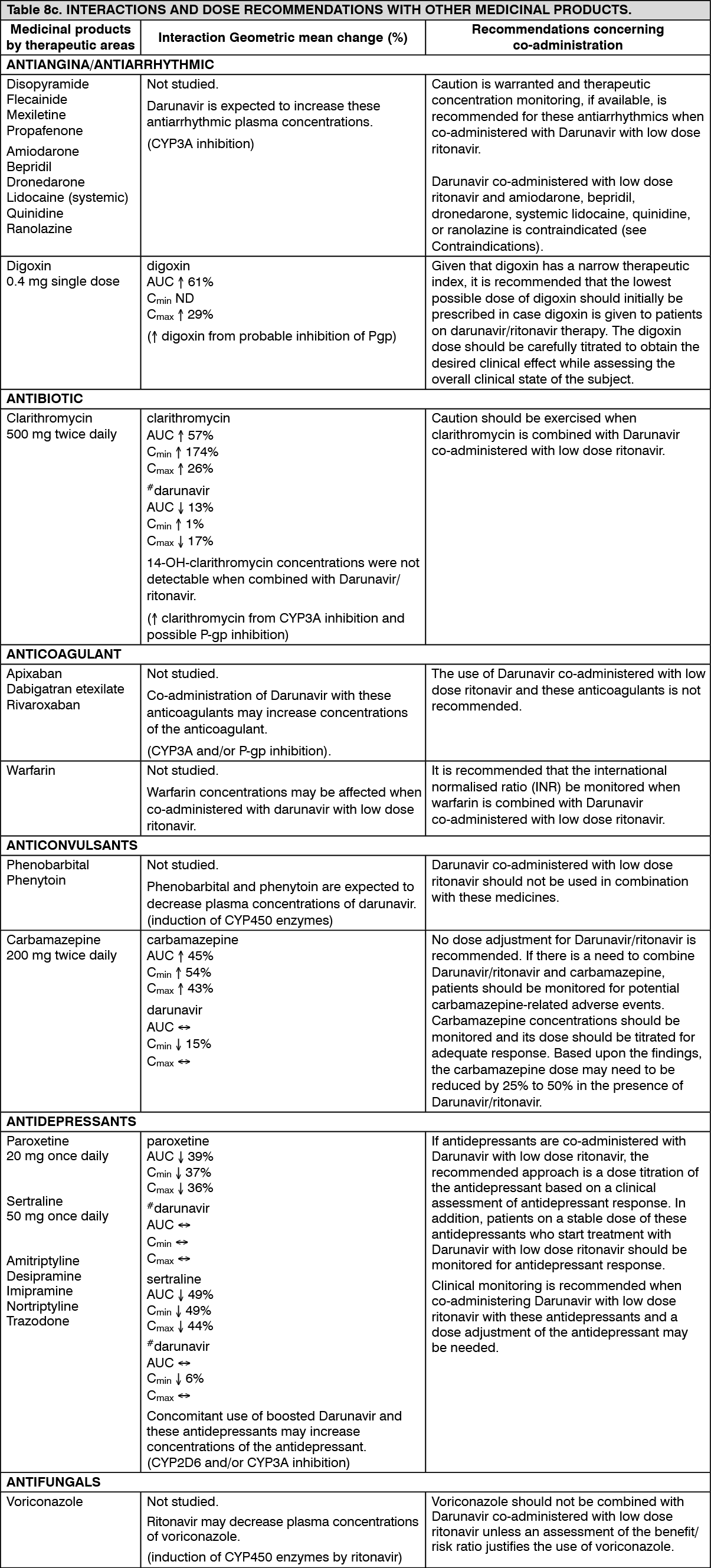 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image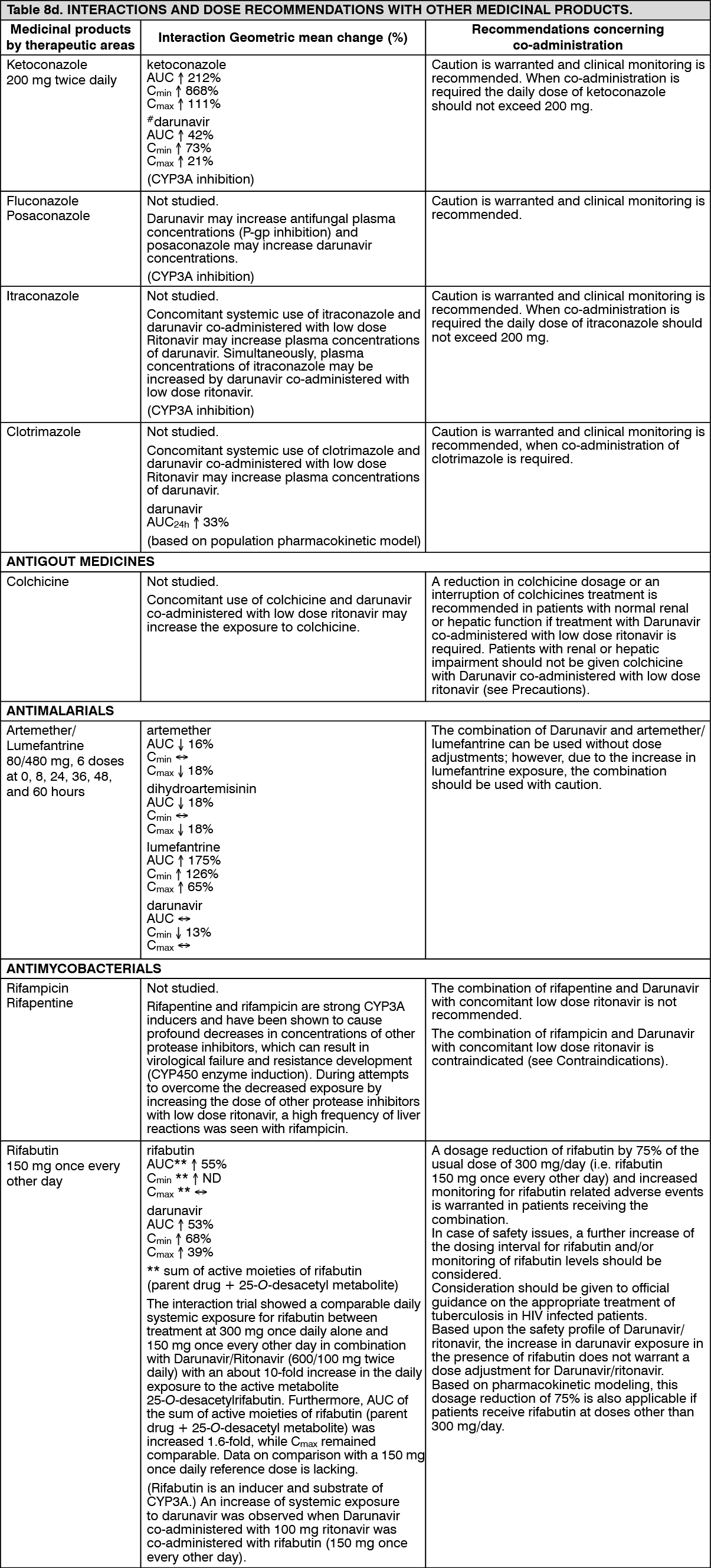 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image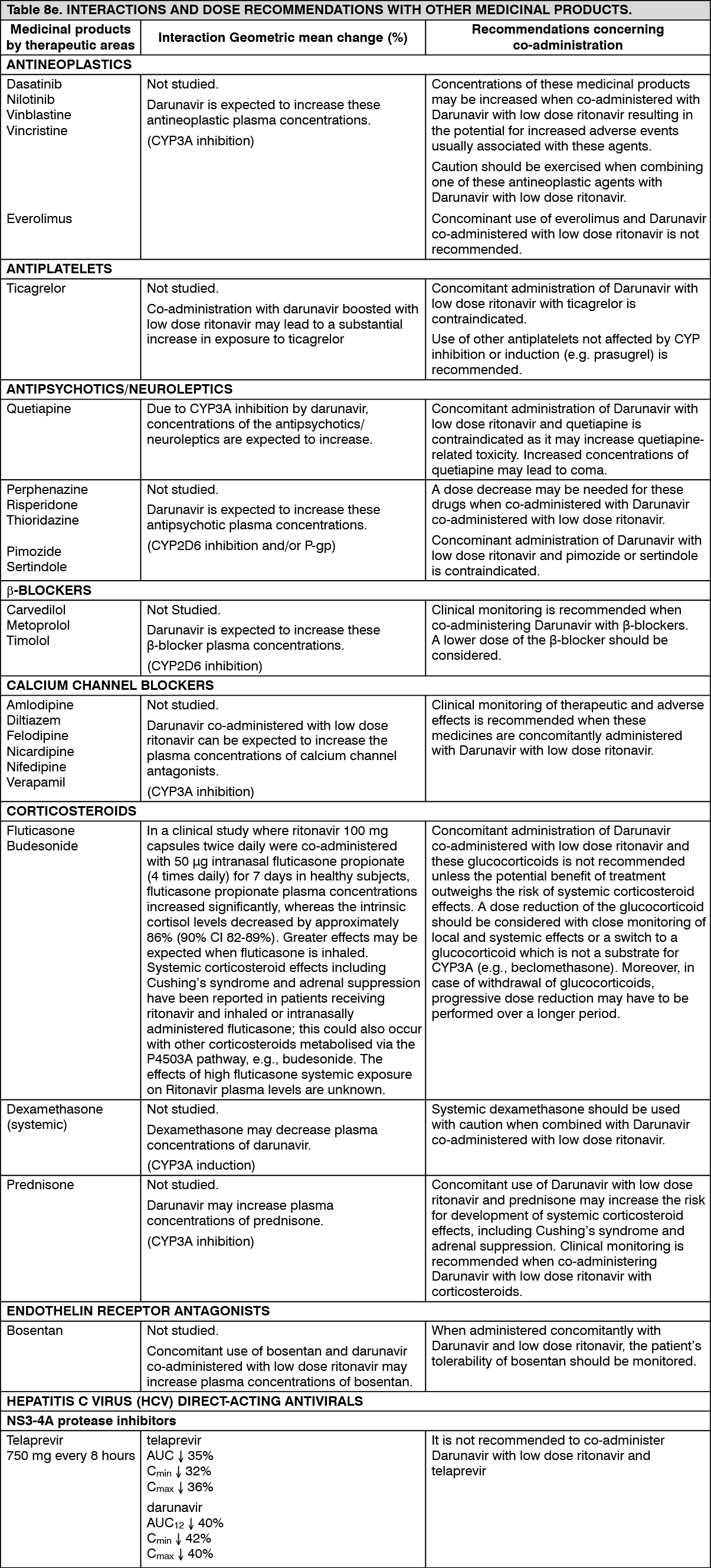 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
