


 Sign Out
Sign Out

In the 96 week analysis, the safety profile of Darunavir/ritonavir 800/100 mg once daily in treatment-naïve subjects was similar to that seen with Darunavir/ritonavir 600/100 mg twice daily in treatment-experienced subjects except for nausea which was observed more frequently in treatment-naïve subjects. This was driven by mild intensity nausea. No new safety findings were identified in the 192 week analysis of the treatment-naïve subjects in which the mean treatment duration of Darunavir/ritonavir 800/100 mg once daily was 162.5 weeks.
Tabulated list of adverse reactions: Adverse reactions are listed by system organ class (SOC) and frequency category. Within each frequency category, adverse reactions are presented in order of decreasing seriousness. Frequency categories are defined as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000) and not known (frequency cannot be estimated from the available data).
Adverse reactions observed with darunavir/ritonavir in clinical trials and post-marketing: See Table 7.
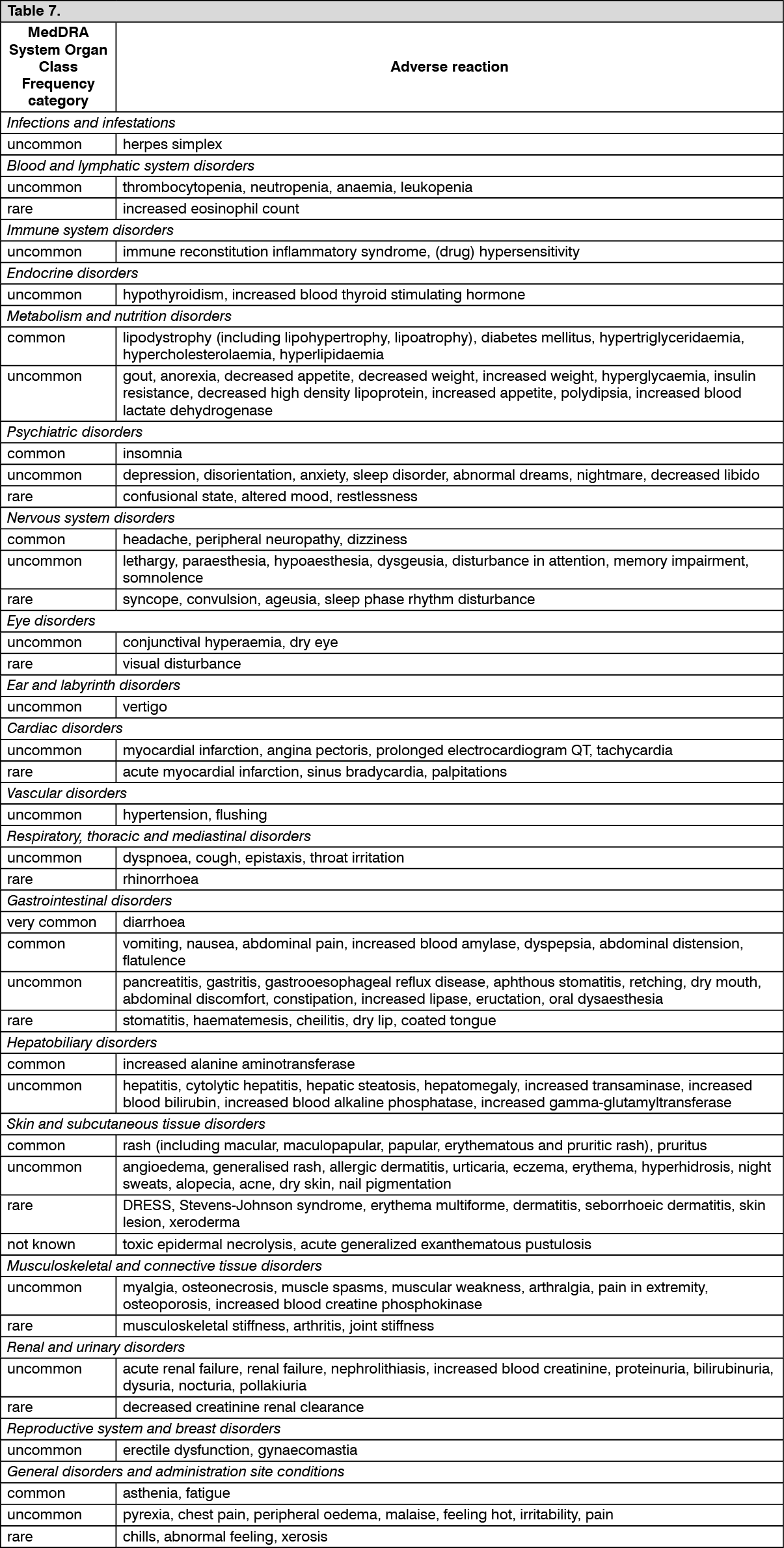 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Rash: In clinical trials, rash was mostly mild to moderate, often occurring within the first four weeks of treatment and resolving with continued dosing. In cases of severe skin reaction see the warning in Precautions.
During the clinical development program of raltegravir in treatment-experienced patients, rash, irrespective of causality, was more commonly observed with regimens containing Darunavir/ritonavir + raltegravir compared to those containing Darunavir/ritonavir without raltegravir or raltegravir without Darunavir/ritonavir. Rash considered by the investigator to be drug-related occurred at similar rates. The exposure-adjusted rates of rash (all causality) were 10.9, 4.2, and 3.8 per 100 patient-years (PYR), respectively; and for drug-related rash were 2.4, 1.1, and 2.3 per 100 PYR, respectively. The rashes observed in clinical studies were mild to moderate in severity and did not result in discontinuation of therapy (see Precautions).
Lipodystrophy: Combination antiretroviral therapy has been associated with redistribution of body fat (lipodystrophy) in HIV patients, including loss of peripheral and facial subcutaneous fat, increased intra-abdominal and visceral fat, breast hypertrophy and dorsocervical fat accumulation (buffalo hump) (see Precautions).
Metabolic abnormalities: Combination antiretroviral therapy has also been associated with metabolic abnormalities such as hypertriglyceridaemia, hypercholesterolaemia, insulin resistance, hyperglycaemia and hyperlactataemia (see Precautions).
Musculoskeletal abnormalities: Increased CPK, myalgia, myositis and rarely, rhabdomyolysis have been reported with the use of protease inhibitors, particularly in combination with NRTIs.
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see Precautions).
Immune reconstitution inflammatory syndrome: In HIV infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see Precautions).
Bleeding in haemophiliac patients: There have been reports of increased spontaneous bleeding in haemophiliac patients receiving antiretroviral protease inhibitors (see Precautions).
Paediatric population: The safety assessment in paediatric patients is based on the 48-week analysis of safety data from three Phase II trials. The following patient populations were evaluated (see Pharmacology: Pharmacodynamics under Actions): 80 ART-experienced HIV-1 infected paediatric patients aged from 6 to 17 years and weighing at least 20 kg who received Darunavir tablets with low dose ritonavir twice daily in combination with other antiretroviral agents.
21 ART-experienced HIV-1 infected paediatric patients aged from 3 to <6 years and weighing 10 kg to <20 kg (16 participants from 15 kg to <20 kg) who received Darunavir oral suspension with low dose ritonavir twice daily in combination with other antiretroviral agents.
12 ART-naïve HIV-1 infected paediatric patients aged from 12 to 17 years and weighing at least 40 kg who received Darunavir tablets with low dose ritonavir once daily in combination with other antiretroviral agents (see Pharmacology: Pharmacodynamics under Actions).
Other special populations: Patients co-infected with hepatitis B and/or hepatitis C virus: Among 1,968 treatment-experienced patients receiving Darunavir co-administered with ritonavir 600/100 mg twice daily, 236 patients were co-infected with hepatitis B or C. Co-infected patients were more likely to have baseline and treatment emergent hepatic transaminase elevations than those without chronic viral hepatitis (see Precautions).
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
View ADR Monitoring Form