Each film coated tablet contains Abiraterone Acetate 250 mg.
Excipients/Inactive Ingredients: Lactose Monohydrate, Microcrystalline Cellulose, Croscarmellose sodium, Povidone, Sodium Lauryl Sulfate, Colloidal Silicon Dioxide, Magnesium Stearate, Opadry White OY-58900 and Purified Water.
Colour: Titanium Dioxide.
Pharmacology: Pharmacodynamics: Mechanism of action: Abiraterone acetate is converted in vivo to abiraterone, an androgen biosynthesis inhibitor. Specifically, abiraterone selectively inhibits the enzyme 17α-hydroxylase/C17, 20-lyase (CYP17). This enzyme is expressed in and is required for androgen biosynthesis in testicular, adrenal and prostatic tumour tissues. CYP17 catalyses the conversion of pregnenolone and progesterone into testosterone precursors, DHEA and androstenedione, respectively, by 17α-hydroxylation and cleavage of the C17, 20 bond. CYP17 inhibition also results in increased mineralocorticoid production by the adrenals.
Androgen-sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with LHRH analogues or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumour. Treatment with abiraterone decreases serum testosterone to undetectable levels (using commercial assays) when given with LHRH analogues (or orchiectomy).
Pharmacodynamics effects: Abiraterone decreases serum testosterone and other androgens to levels lower than those achieved by the use of LHRH analogues alone or by orchiectomy. This results from the selective inhibition of the CYP17 enzyme required for androgen biosynthesis. Prostate specific antigen (PSA) serves as a biomarker in patients with prostate cancer. In a phase 3 clinical study of patients who failed prior chemotherapy with taxanes, 38% of patients treated with abiraterone acetate, versus 10% of patients treated with placebo, had at least a 50% decline from baseline in PSA levels.
Pharmacokinetics: Following administration of abiraterone acetate, the pharmacokinetics of abiraterone and abiraterone acetate have been studied in healthy subjects, patients with metastatic advanced prostate cancer and subjects without cancer with hepatic or renal impairment. Abiraterone acetate is rapidly converted in vivo to abiraterone, an androgen biosynthesis inhibitor.
Absorption: Following oral administration of abiraterone acetate in the fasting state, the time to reach maximum plasma abiraterone concentration is approximately 2 hours.
Administration of abiraterone acetate with food, compared with administration in a fasted state, results in up to a 10-fold (AUC) and up to a 17-fold (Cmax) increase in mean systemic exposure of abiraterone, depending on the fat content of the meal. Given the normal variation in the content and composition of meals, taking abiraterone with meals has the potential to result in highly variable exposures. Therefore, abiraterone must not be taken with food. It should be taken at least two hours after eating and no food should be eaten for at least one hour after taking abiraterone. The tablets should be swallowed whole with water.
Distribution: The plasma protein binding of 14C-abiraterone in human plasma is 99.8%. The apparent volume of distribution is approximately 5,630 L suggesting that abiraterone extensively distributes to peripheral issues.
Biotransformation: Following oral administration of 14C-abiraterone acetate as capsules, abiraterone acetate is hydrolysed to abiraterone, which then undergoes metabolism including sulphation, hydroxylation and oxidation primarily in the liver. The majority of circulating radioactivity (approximately 92%) is found in the form of metabolites of abiraterone. Of 15 detectable metabolites 2 main metabolites, abiraterone sulphate and N-oxide abiraterone sulphate, each represents approximately 43% of total radioactivity.
Elimination: The mean half-life of abiraterone in plasma is approximately 15 hours based on data from healthy subjects. Following oral administration of 14C-abiraterone acetate 1,000 mg, approximately 88% of the radioactive dose is recovered in faeces and approximately 5% in urine. The major compounds present in faeces are unchanged abiraterone acetate and abiraterone (approximately 55% and 22% of the administered dose, respectively).
Patients with hepatic impairment: The pharmacokinetics of abiraterone acetate was examined in subjects with pre-existing mild or moderate hepatic impairment (Child-Pugh Class A and B, respectively, and in healthy control subjects. Systemic exposure to abiraterone after a single oral 1,000 mg dose increased by approximately 11% and 260% in subjects with mild and moderate pre-existing hepatic impairment, respectively. The mean half-life of abiraterone is prolonged to approximately 18 hours in subjects with mild hepatic impairment and to approximately 19 hours in subjects with moderate hepatic impairment.
In another trial, the pharmacokinetics of abiraterone were examined in subjects with pre-existing severe (n=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The AUC to abiraterone increased by approximately 600% and the fraction of free drug increased by 80% in subjects with severe hepatic impairment compared to subjects with normal hepatic function.
No dose adjustment is necessary for patients with pre-existing mild hepatic impairment.
The use of abiraterone acetate should be cautiously assessed in patients with moderate hepatic impairment in whom the benefit clearly should outweigh the possible risk. Abiraterone acetate should not be used in patients with severe hepatic impairment.
For patients who develop hepatotoxicity during treatment, suspension of treatment and dose adjustment may be required.
Patients with renal impairment: The pharmacokinetics of abiraterone acetate was compared in patients with end-stage renal disease on a stable haemodialysis schedule versus matched control subjects with normal renal function. Systemic exposure to abiraterone after a single oral 1,000 mg dose did not increase in subjects with end-stage renal disease on dialysis.
Administration in patients with renal impairment, including severe renal impairment, does not require dose reduction. However, there is no clinical experience in patients with prostate cancer and severe renal impairment. Caution is advised in these patients.
Abiraterone is indicated with prednisone or prednisolone for: The treatment of newly diagnosed high risk metastatic hormone sensitive prostate cancer (mHSPC) in adult men in combination with androgen deprivation therapy (ADT).
The treatment of metastatic castration resistant prostate cancer (mCRPC) in adult men who are asymptomatic or mildly Symptomatic after failure of androgen deprivation therapy in whom chemotherapy is not yet clinically indicated.
The treatment of metastatic castration resistant prostate cancer (mCRPC) in adult men whose disease has progressed on or after a docetaxel-based chemotherapy regimen.
Posology: Recommended dose for high risk metastatic HSPC: The recommended dose of abiraterone is 1,000 mg (four 250 mg tablets) orally once daily with prednisone 5 mg administration orally once daily. (See information on the Method of administration as follows).
Recommended dose for metastatic CRPC: The recommended dose is 1,000 mg (four 250 mg tablets) orally as a single daily dose (see information on Method of administration as follows). Abiraterone is to be taken with low dose prednisone or prednisolone. The recommended dose of prednisone or prednisolone is 10 mg daily.
Medical castration with LHRH analogue should be continued during treatment in patients not surgically castrated. Recommended monitoring serum transaminases should be measured prior to starting treatment, every two weeks for the first three months of treatment and monthly thereafter. Blood pressure, serum potassium and fluid retention should be monitored monthly. However, patients with a significant risk for congestive heart failure should be monitored every 2 weeks for the first three months of treatment and monthly thereafter.
In patients with pre-existing hypokalaemia or those that develop hypokalaemia whilst being treated with abiraterone, consider maintaining the patient's potassium level at ≥ 4.0 mM.
For patients who develop Grade ≥ 3 toxicities including hypertension, hypokalaema, oedema and other non-mineralocorticoid toxicities, treatment should be withheld and appropriate medical management should be instituted. Treatment with abiraterone should not be reinitiated until symptoms of the toxicity have resolved to Grade 1 or baseline.
In the event of a missed daily dose of either abiraterone, prednisone or prednisolone, treatment should be resumed the following day with the usual daily dose.
Hepatotoxicity: For patients who develop hepatotoxicity during treatment (alanine aminotransferase [ALT] increases or aspartate aminotransferase [AST] increases above 5 times the upper limit of normal ULN]), treatment should be withheld immediately. Re-treatment following return of liver function tests to the patient's baseline may be given at a reduced dose of 500 mg (two tablets) once daily. For patients being re-treated, serum transaminases should be monitored at a minimum of every two weeks for three months and monthly thereafter. If hepatotoxicity recurs at the reduced dose of 500 mg daily treatment should be discontinued.
If patients develop severe hepatotoxicity (ALT or AST 20 times the upper limit of normal) anytime while on therapy, treatment should be discontinued and patients should not be re-treated.
Hepatic function impairment: No dose adjustment is necessary for patients with pre-existing mild hepatic impaired. Child-Pugh Class A.
Moderate hepatic impairment (Child-Pugh Class B) has been shown to increase the systemic exposure to abiraterone by approximately four-fold following single oral doses of abiraterone acetate 1,000 mg. There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). No dose adjustment can be predicted. The use of abiraterone should be cautiously assessed in patients with moderate hepatic impairment, in whom the benefit clearly should outweigh the possible risk. Abiraterone should not be used in patents with severe hepatic impairment.
Renal impairment: No dose adjustment is necessary for patients with renal impairment. However, there is no clinical experience in patients with prostate cancer and severe renal impairment. Caution is advised in these patients.
Paediatric population: There is no relevant use of abiraterone in the paediatric population.
Method of administration: Abiraterone is for oral use. Abiraterone should be taken at least two hours after eating and no food should be eaten for at least one hour after taking the tablets. These should be swallowed whole with water.
Human experience of overdose with abiraterone is limited.
There is no specific antidote. In the event of an overdose, administration should be withheld and general supportive measures undertaken, including monitoring for arrhythmias, hypokalaemia and for signs and symptoms of fluid retention. Liver function also should be assessed.
Hypersensitivity to the active substance or to any of the excipients.
Women who are or may potentially be pregnant.
Severe hepatic impairment (Child-Pugh Class C).
Hypertension, hypokalemia, fluid retention and cardiac failure due to mineralocorticoid excess: Abiraterone may cause hypertension, hypokalemia and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition. Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in incidence and severity of these adverse reactions. Caution is required in treating patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia (e.g., those on cardiac glycosides), or fluid retention (e.g., those with heart failure, severe or unstable angina pectoris, recent myocardial infarction or ventricular arrhythmia and those with severe renal impairment).
Abiraterone should be used with caution in patients with a history of cardiovascular disease. The phase 3 studies conducted with abiraterone excluded patients with uncontrolled hypertension, clinically significant heart disease as evidenced by myocardial infarction, or arterial thrombotic events in the past 6 months, severe or unstable angina, or New York Heart Association Class (NYHA) III or IV heart failure (in study 301) a Class II to IV heart failure (in study 302) or cardiac ejection fraction measurement of <50%. In study 302 patients with atrial fibrillation, or other cardiac arrhythmia requiring medical therapy were excluded. Safety in patients with left ventricular ejection fraction (LVEF <50% or NYHA Class III or IV heart failure (in study 301) or NYHA Class II to IV heart failure (in study 302) was not established.
Before treating patients with a significant risk for congestive heart failure (e.g. a history of cardiac failure, uncontrolled hypertension, or cardiac events such as ischaemic heart disease), consider obtaining an assessment of cardiac function (e.g., echocardiogram). Before treatment with abiraterone, cardiac failure should be treated and cardiac function optimised. Hypertension, hypokalaemia and fluid retention should be corrected and controlled. During treatment, blood pressure, serum potassium, fluid retention (weight gain, peripheral oedema) and other signs and symptoms of congestive heart failure should be monitored every 2 weeks for 3 months, then monthly thereafter and abnormalities corrected. QT prolongation has been observed in patients experiencing hypokalemia in association with abiraterone treatment. Assess cardiac function as clinically indicated, institute appropriate management and consider discontinuation of this treatment if there is a clinically significant decrease in cardiac function.
Hepatotoxicity and hepatic impairment: Marked increases in liver enzymes leading to treatment discontinuation or dose modification occurred in controlled clinical studies. Serum transaminase levels should be measured prior to starting treatment, every two weeks for the first three months of treatment, and monthly thereafter. If clinical symptoms or signs suggestive of hepatotoxicity develop, serum transaminases should be measured immediately. If at any time the ALT or AST rises above 5 times the upper limit of normal (ULN), treatment should be interrupted immediately and liver function closely monitored. Re-treatment may take place only after return of liver function tests to the patient's baseline and at a reduced dose level.
If patients develop severe hepatotoxicity (ALT or AST 20 times the ULN) anytime while on therapy, treatment should be discontinued and patients should not be re-treated.
Patients with active or symptomatic viral hepatitis were excluded from clinical trials; thus, there are no data to support the use of abiraterone in this population.
There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). The use of abiraterone should be cautiously assessed in patients with moderate hepatic impairment, in whom the benefit clearly should outweigh the possible risk.
Abiraterone should not be used in patients with severe hepatic impairment.
There have been rare post-marketing reports of acute liver failure and hepatitis fulminant some with fatal outcome.
Corticosteroid withdrawal and coverage of stress situations: Caution is advised and monitoring for adrenocortical insufficiency should occur if patients are withdrawn from prednisone or prednisolone. If abiraterone is continued after corticosteroids are withdrawn, patients should be monitored for symptoms of mineralocorticoid excess.
In patients on prednisone or prednisolone who are subjected to unusual stress, an increased dose of corticosteroids may be indicated before, during and after the stressful situation.
Bone density: Decreased bone density may occur in men with metastatic advanced prostate cancer (castration resistant prostate cancer). The use of abiraterone in combination with a glucocorticoid could increase this effect.
Prior use of ketoconazole: Lower rates of response might be expected in patients previously treated with ketoconazole for prostate cancer.
Hyperglycemia: The use of glucocorticoids could increase hyperglycemia, therefore blood sugar should be measured frequently in patients with diabetes.
Use with chemotherapy: The safety and efficacy of concomitant use of abiraterone with cytotoxic chemotherapy has not been established.
Potential risks: Anemia and sexual dysfunction may occur in men with metastatic castration resistant prostate cancer including those undergoing treatment with abiraterone.
Skeletal muscle effects: Cases of myopathy have been reported in patients treated with abiraterone. Some patients had rhabdomyolysis with renal failure. Most cases developed within the first month of treatment and recovered after abiraterone withdrawal. Caution is recommended in patients concomitantly treated with drugs known to be associated with myopathy/rhabdomyolysis.
Interactions with other medicinal products: Strong inducers of CYP3A4 during treatment are to be avoided unless there is no therapeutic alternative, due to risk of decreased exposure to abiraterone.
Effects on ability to drive and use machines: Abiraterone has no or negligible influence on the ability to drive or use machines.
Women of childbearing potential: There are no human data on the use of abiraterone in pregnancy and this medicinal product is not for use in women of childbearing potential.
Contraception in males and females: It is not known whether abiraterone or its metabolites are present in semen. A condom is required if the patient is engaged in sexual activity with a pregnant woman. If the patient is engaged in sex with a woman of childbearing potential, a condom is required along with another effective contraceptive method. Studies in animals have shown reproductive toxicity.
Pregnancy: Abiraterone is not for use in women and is contraindicated in women who are or may potentially be pregnant.
Breast-feeding: Abiraterone is not for use in women.
Fertility: Abiraterone affected fertility in male and female rats, but these effects were fully reversible.
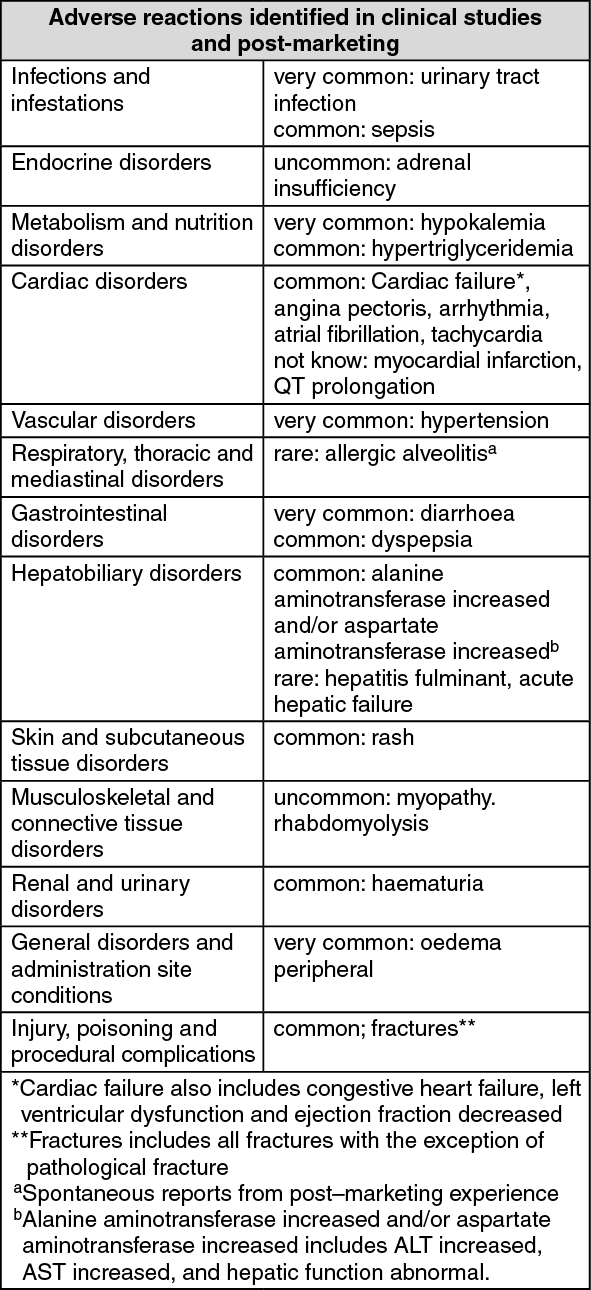
Summary of the safety profile: The most common adverse reactions seen are peripheral oedema, hypokalemia, hypertension and urinary tract infection. Other important adverse reactions include, cardiac disorders, hepatotoxicity, fractures, and allergic alveolitis.
Abiraterone may cause hypertension, hypokalemia and fluid retention as a pharmacodynamics consequence of its mechanism of action. In clinical studies, anticipated mineralocorticoid adverse reactions were seen more commonly in patients treated with abiraterone acetate than in patients treated with placebo: hypokalemia 21% versus 11%, hypertension 16% versus 11% and fluid retention (peripheral oedema) 26% versus 20%, respectively. In patients treated with abiraterone acetate, CTCAE (version 3.0) Grades 3 and 4 hypokalemia and CTCAE (version 3.0) Grades 3 and 4 hypertension were observed in 4% and 2% of patients, respectively. Mineralocorticoid reactions generally were able to be successfully managed medically. Concomitant use of a corticosteroid reduces the incidence and severity of these adverse reactions.
Tabulated summary of adverse reactions: In studies of patients with metastatic advanced prostate cancer who were using a luteinising hormone-releasing hormone (LHRH) analogue, or were previously treated with orchiectomy, abiraterone was administered at a dose of 1,000 mg daily in combination with low dose prednisone or prednisolone (either 5 or 10 mg daily depending on the indication).
Adverse reactions observed during clinical studies and post-marketing experience are listed as follows by frequency category. Frequency categories are defined as follows: very common ( ≥1/10): common (≥ 1/100 to < 1/10): uncommon ( ≥ 1/1,000 to <1/100); rare ( ≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000) and not known (frequency cannot be estimated from the available data).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The following CTCAE (version 3.0) Grade 3 adverse reactions occurred in patients treated with abiraterone acetate: hypokalemia 3%; urinary tract infection, alanine aminotransferase increased, hypertension, aspartate aminotransferase increased, fractures 2%; peripheral oedema, cardiac failure, and atrial fibrillation 1% each. CTCAE (version 3.0) Grade 3 hypertri-glyceridemia and angina pectoris occurred in < 1% of patients. CTCAE (version 3.0) Grade 4 peripheral oedema, hypokalemia, urinary tract infection, cardiac failure and fractures occurred in <1% of patients.
Description of selected adverse reactions: Cardiovascular reactions: Both phase 3 studies excluded patients with uncontrolled hypertension, clinically significant heart disease as evidenced by myocardial infarction, or arterial thrombotic events in the past 6 months, severe or unstable angina, or NYHA Class III or IV heart failure (study 301) or Class II to IV heart failure (study 302) or cardiac ejection fraction measurement of < 50%. All patients enrolled (both active and placebo-treated patients) were concomitantly treated with androgen deprivation therapy, predominantly with the use of LHRH analogues, which has been associated with diabetes, myocardial infarction, cerebrovascular accident and sudden cardiac death. The incidence of cardiovascular adverse reactions in the phase 3 studies in patients taking abiraterone acetate versus patients taking placebo were as follows: hypertension 14.5% VS. 10.5%, atrial fibrillation 3.4% vs. 3.4%, tachycardia 2.8% vs. 1.7%, angina pectoris 1.9% vs. 0.9%, cardiac failure 1.9% vs. 0.6%, and arrhythmia 1.1% vs. 0.4%.
Hepatotoxicity: Hepatotoxicity with elevated ALT, aspartate transaminase (AST) and total bilirubin has been reported in patients treated with abiraterone acetate. Across all clinical studies, liver function test elevations (ALT or AST increases of > 5x ULN or bilirubin increases > 1.5x ULN) were reported in approximately 4% of patients who received abiraterone acetate, typically during the first 3 months after starting treatment. In the 301 clinical study, patients whose baseline ALT or AST were elevated were more likely to experience liver function test elevations than those beginning with normal values. When elevations of either ALT or AST > 5x ULN, or elevations in bilirubin > 3x ULN were observed, abiraterone acetate was withheld or discontinued. In two instances marked increases in liver function tests occurred. These two patients with normal baseline hepatic function, experienced ALT or AST elevations 15 to 40x ULN and bilirubin elevations 2 to 6x ULN. Upon discontinuation of treatment, both patients had normalisation of their liver function tests and one patient was re-treated without recurrence of the elevations. In study 302, Grade 3 or 4 ALT or AST elevations were observed in 35 (6.5%) patients treated with abiraterone acetate. Aminotransferase elevations resolved in all but 3 patients (2 with new multiple liver metastases and 1 with AST elevation approximately 3 weeks after the last dose of abiraterone acetate). Treatment discontinuations due to ALT and AST increases were reported in 1.7% and 1.3% of patients treated with abiraterone acetate and 0.2% and 0% of patients treated with placebo, respectively. No deaths were reported due to hepatotoxicity event.
In clinical trials, the risk for hepatotoxicity was mitigated by exclusion of patients with baseline hepatitis or significant abnormalities of liver function tests. In the 301 trial, patients with baseline ALT and AST ≥ 2.5x ULN in the absence of liver metastases and > 5x ULN in the presence of liver metastases were excluded. In the 302 trial, patients with liver metastases were not eligible and patients with baseline ALT and AST ≥ 2.5x ULN were excluded. Abnormal liver function tests developing in patients participating in clinical trials were vigorously managed by requiring treatment interruption and permitting re-treatment only after return of liver function tests to the patient's baseline. Patients with elevations of ALT or AST > 20x ULN were not re-treated. The safety of re-treatment in such patients is unknown. The mechanism for hepatotoxicity is not understood.
Effect of food on abiraterone acetate: Administration with food significantly increases the absorption of abiraterone acetate. The efficacy and safety when given with food have not been established therefore this medicinal product must not be taken with food.
Potential for other medicinal products to affect abiraterone exposures: In a clinical pharmacokinetic interaction study of healthy subjects pretreated with a strong CYP3A4 inducer rifampicin, 600 mg daily for 6 days followed by a single dose of abiraterone acetate 1,000 mg, the mean plasma AUC∞ of abiraterone was decreased by 55%.
Strong inducers of CYP3A4 (e.g., phenytoin, carbamazepine, rifampicin, rifabutin, rifapentine, phenobarbital, St John's wort [Hypericum perforatum]) during treatment are to be avoided, unless there is no therapeutic alterative.
In a separate clinical pharmacokinetic interaction study of healthy subjects, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone.
Potential to affect exposures to other medicinal products: Abiraterone is an inhibitor of the hepatic drug-metabolizing enzymes CYP2D6 and CYP2CB.
In a study to determine the effects of abiraterone acetate (plus prednisone) on a single dose of the CYP2D6 substrate dextromethorphan, the systemic exposure (AUC) of dextromethorphan was increased approximately 2.9 fold. The AUC24 for dextrorphan, the active metabolite of dextromethorphan, increased approximately 33%.
Caution is advised when administering with medicinal products activated by a metabolised by CYP2D6 particularly with medicinal products that have a narrow therapeutic index. Dose reduction of medicinal products with a narrow therapeutic index that are metabolised by CYP2D6 should be considered. Examples of medicinal products metabolised by CYP2D6 include metoprolol, propranolol, desipramine, venlafaxine, haloperidol, risperidone, propatenone, flecainide, codeine, oxycodone and tramadol (the latter three products requiring CYP2D6 to form their active analgesic metabolites).
In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone was increased by 46% and the AUCs for M-III and M-IV, the active metabolites of pioglitazone, each decreased by 10% when pioglitazone was given together with a single dose of 1,000 mg abiraterone acetate. Although these results indicate that no clinically meaningful increases in exposure are expected when abiraterone is combined with drugs that are predominantly eliminated by CYP2C8, patients should be monitored for signs of toxicity related to a CYP2C8 substrate with a narrow therapeutic index if used concomitantly.
In vitro, the major metabolites abiraterone sulphate and N-oxide abiraterone sulphate were shown to inhibit the hepatic uptake transporter OATP1B1 and as a consequence it may increase the concentrations of drugs eliminated by OATP1B1. There are no clinical data available to confirm transporter based interaction.
Use with products known to prolong QT interval: Since androgen deprivation treatment may prolong the QT interval, caution is advised when administering abiraterone with medicinal products known to prolong the QT interval or medicinal products able to induce torsades de pointes such as class IA (e.g. quinidine, disopyramide) or class III (e.g. amiodarone, sotalol, dofetilide, butilide) antiarrhythmic medicinal products, methadone, moxifloxacin, antipsychotics, etc.
Use with Spironolactone: Spironolactone binds to the androgen receptor and may increase prostate stearic antigen (PSA) levels. Use with abiraterone is not recommended.
Shelf-Life: 36 months from the date of manufacture.
Store below 30°C.
Protect from light and moisture.
L02BX03 - abiraterone ; Belongs to the class of other hormone antagonists and related agents. Used in the treatment of metastatic castration-resistant prostate cancer.
Abiratred FC tab 250 mg
120's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
