For Intravenous Use After Reconstitution: Treatment with XYNTHA Antihemophilic Factor (Recombinant), Plasma/Albumin-Free should be initiated under the supervision of a physician experienced in the treatment of hemophilia A.
Dosage and duration of treatment depend on the severity of the factor VIII deficiency, the location and extent of bleeding, and the patient's clinical condition. Doses administered should be titrated to the patient's clinical response. In the presence of an inhibitor, higher doses may be required.
One International Unit (IU) of factor VIII activity corresponds approximately to the quantity of factor VIII in one milliliter (mL) of normal human plasma. The calculation of the required dosage of factor VIII is based upon the empirical finding that, on average, 1 IU of factor VIII per kg body weight raises the plasma factor VIII activity by approximately 2 IU/dL. The required dosage is determined using the following formula: The expected
in vivo peak increase in factor VIII level expressed as IU/dL (or % normal) can be estimated using the following formulas: (See equation.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The labeled potency of XYNTHA is based on the European Pharmacopoeia chromogenic substrate assay, in which the Wyeth manufacturing standard has been calibrated using a one-stage clotting assay. This method of potency assignment is intended to harmonize XYNTHA with clinical monitoring using a one-stage clotting assay.
Control of Bleeding Episodes: In the case of the following bleeding events, consideration should be given to maintaining the factor VIII activity at or above the plasma levels (in % of normal or in IU/dL) outlined as follows for the indicated period.
The following chart can be used to guide dosing in bleeding episodes: (See Table 5.)
Click on icon to see table/diagram/image
Routine and Surgical Prophylaxis in Patients with Hemophilia A: In the case of the following bleeding events, consideration should be given to maintaining the factor VIII activity at or above the plasma levels (in % of normal or in IU/dL) outlined as follows for the indicated period. Monitoring of replacement therapy by means of plasma factor VIII activity is recommended, particularly for surgical intervention.
The following chart can be used to guide dosing in surgery: (See Table 6.)
Click on icon to see table/diagram/image
Dosage for prophylaxis: XYNTHA has been administered prophylactically in a pivotal clinical trial in previously treated adolescent and adults at a dose of 30 ± 5 IU/kg given 3 times weekly.
The recommended starting regimen in children (<12 years) is 25 ± 5 IU/kg of XYNTHA administered every other day. More frequent or higher doses may be required in children <12 years of age to account for the higher clearance in this age group (see Pharmacology: Pharmacodynamics under Actions).
Adjust the dosing regimen (dose or frequency) based on the patient's clinical response.
Instructions for Use: XYNTHA is administered by IV infusion after reconstitution of the freeze-dried powder with the supplied pre-filled diluent (0.9% Sodium Chloride solution) syringe.
Patients should follow the specific reconstitution and administration procedures provided by their physicians. The procedures as follows are provided as general guidelines for the reconstitution and administration of XYNTHA.
Preparation and Reconstitution: Preparation: 1. Always wash hands before performing the following procedures.
2. Aseptic technique (meaning clean and germ-free) should be used during the reconstitution procedure.
3. All components used in the reconstitution and administration of this product should be used as soon as possible after opening the sterile containers, to minimize unnecessary exposure to the atmosphere.
Note: If the patient uses more than one vial of XYNTHA per infusion, each vial should be reconstituted as per the following instructions. The diluent syringe should be removed, leaving the vial adapter in place, and a separate, single large luer lock syringe may be used to draw back the reconstituted contents of each of the individual vials. Do not detach the diluent syringes or the large luer lock syringe until the patient is ready to attach the large luer lock syringe to the next vial adapter.
Reconstitution: 1. Allow the vials of freeze-dried XYNTHA and the pre-filled diluent syringe to reach room temperature.
2. Remove the plastic flip-top cap from the XYNTHA vial to expose the central portions of the rubber stopper.
3. Wipe the top of the vial with the alcohol swab provided, or use another antiseptic solution, and allow to dry. After cleaning, do not touch the rubber stopper with the hand or allow it to touch any surface.
4. Peel back the cover from the clear plastic vial adapter package.
Do not remove the adapter from the package.
5. Place the vial on a flat surface. While holding the adapter package, place the vial adapter over the vial and press down firmly on the package until the adapter spike penetrates the vial stopper.
6. Grasp the plunger rod. Avoid contact with the shaft of the plunger rod. Attach the threaded end of the plunger rod to the diluent syringe plunger by pushing and turning firmly.
7. Break off the tamper-resistant plastic tip cap from the diluent syringe by snapping the perforation of the cap. Do not touch the inside of the cap or the syringe tip. The diluent syringe may need to be recapped (if not administering reconstituted XYNTHA immediately), so place the cap on its top on a clean surface in a spot where it would be least likely to become environmentally contaminated.
8. Lift the package away from the adapter and discard the package.
9. Place the vial on a flat surface. Connect the diluent syringe to the vial adapter by inserting the tip into the adapter opening while firmly pushing and turning the syringe clockwise until the connection is secured.
10. Slowly depress the plunger rod to inject all the diluent into the XYNTHA vial.
11. Without removing the syringe,
gently swirl the contents of the vial until the powder is dissolved.
Note: The final solution should be inspected visually for particulate matter before administration. The solution should be clear to slightly opalescent and colorless. If it is not, the solution should be discarded and a new kit should be used.
12. Invert the vial and slowly draw the solution into the syringe.
13. Detach the syringe from the vial adapter by gently pulling and turning the syringe counter-clockwise. Discard the vial with the adapter attached.
Note: If the solution is not to be used immediately, the syringe cap should be carefully replaced. Do not touch the syringe tip or the inside of the cap.
The reconstituted solution may be stored at room temperature prior to administration, but should be administered within 3 hours of reconstitution.
XYNTHA, when reconstituted, contains polysorbate-80, which is known to increase the rate of di-(2-ethylhexyl)phthalate (DEHP) extraction from polyvinyl chloride (PVC). This should be considered during the preparation and administration of XYNTHA, including storage time elapsed in a PVC container following reconstitution. It is important that the recommendations in Dosage & Administration be followed closely.
Administration: XYNTHA is administered by IV infusion after reconstitution with the supplied pre-filled diluent (0.9% Sodium Chloride solution, 4 mL) syringe. Parenteral drug products should be inspected for particulate matter and discoloration prior to administration, whenever solution and container permit.
XYNTHA should be administered using the infusion set provided in this kit and the pre-filled diluent syringe provided, or a single sterile disposable plastic syringe. In addition, the solution should be withdrawn from the vial using the vial adapter.
1. Attach the syringe to the luer end of the infusion set tubing provided.
2. Apply a tourniquet and prepare the injections site by wiping the skin well with an alcohol swab provided in the kit.
3. Remove the protective needle cover and perform venipuncture. Insert the needle on the infusion set tubing into the vein, and remove the tourniquet. The reconstituted XYNTHA product should be injected intravenously over several minutes. The rate of administration should be determined by the patient's comfort level. As with any intravenous administration, always verify proper needle placement.
Reconstituted XYNTHA should not be administered in the same tubing or container with other medicinal products.
After infusing XYNTHA, remove the infusion set and discard. The amount of drug product left in the infusion set will not affect treatment.
Note: Dispose of all unused solution, the empty vial(s), and other used medical supplies in an appropriate container for throwing away medical waste that might hurt others if not handled properly.
Pediatric population: Safety of XYNTHA was studied in previously treated children and adolescents (n=18, 12-16
years of age in a pivotal study and n=49, 7-16 years of age in a supporting study). In a pivotal study, adverse event data from patients who were ≤16 years of age were compared with data from those over 16 years of age. Eighteen (18) patients were ≤16 years of age and 76 were >16 years of age. Extent of exposure was similar for patients in the two age groups. Treatment emergent adverse events were similar in severity and incidence in the two age groups.
XYNTHA may be used in the same manner as predecessor product ReFacto, because it is biochemically comparable to predecessor product ReFacto and has demonstrated similar pharmacokinetic characteristics with predecessor product ReFacto. Safety and efficacy of predecessor product ReFacto has been studied both in previously treated children and adolescents (n=31, 5-18 years of age) and in previously untreated neonates, infants, and children (n=101, ages <1-52 months). Clinical data derived from completed studies with moroctocog alfa (AF-CC) in PTP (ReFacto AF*: n=37, 18 patients <6 and 19 patients 6 to <12 years of age; XYNTHA: n=51, 46 patients <6 and 5 patients 6 to <16 years of age) and PUP (ReFacto AF: n=23 patients <6 years of age) demonstrated a safety profile similar to that of the predecessor product ReFacto. See also Pharmacology: Pharmacokinetics under Actions.
* ReFacto AF is another moroctocog alfa (AF-CC) product approved in Europe where the labeled potency is based on the European Pharmacopoeia chromogenic substrate assay, in which the manufacturing potency standard has been calibrated to the WHO 8
th International Standard (IS) using the chromogenic substrate assay, while the XYNTHA potency assignment is aligned to the WHO 8
th IS using a one-stage clotting assay.
Elderly population: Clinical studies of XYNTHA did not include subjects 65 years of age and over. In general, dose selection for an elderly patient should be individualized.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image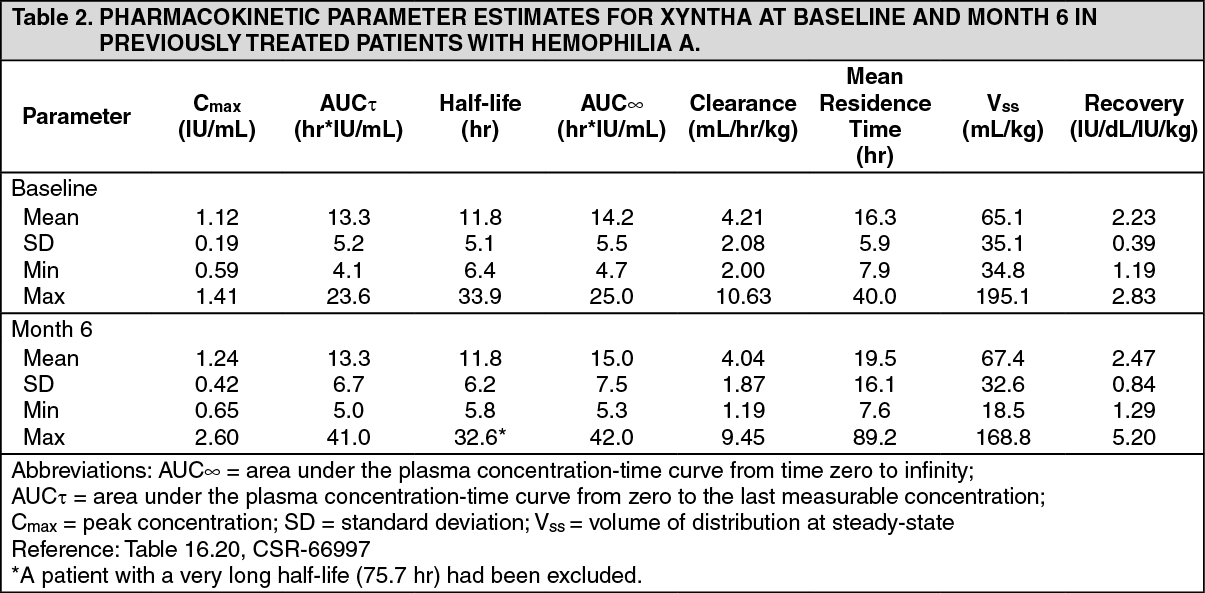 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image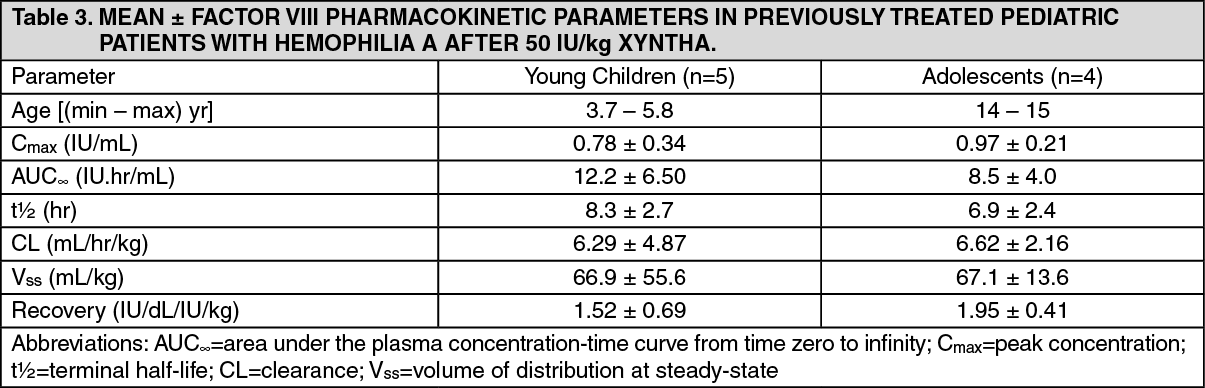 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
