


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of Action: NSCLC: Nintedanib is a triple angiokinase inhibitor blocking vascular endothelial growth factor receptors (VEGFR 1-3), platelet-derived growth factor receptors (PDGFR α and β) and fibroblast growth factor receptors (FGFR 1-3) kinase activity. Nintedanib binds competitively to the adenosine triphosphate (ATP) binding pocket of these receptors and blocks the intracellular signalling which is crucial for the proliferation and survival of endothelial as well as perivascular cells (pericytes and vascular smooth muscle cells). In addition Fms-like tyrosine-protein kinase (Flt)-3, lymphocyte-specific tyrosine-protein kinase (Lck) and proto-oncogene tyrosine-protein kinase Src (Src) are inhibited.
IPF/chronic fibrosing ILDs with a progressive phenotype/SSc-ILD: Nintedanib is a small molecule tyrosine kinase inhibitor including the receptors platelet-derived growth factor receptor (PDGFR) α and β, fibroblast growth factor receptor (FGFR) 1-3, and vascular endothelial growth factor receptor (VEGFR) 1-3. In addition, nintedanib inhibits Lck, Lyn, Src, and CSF1R kinases. Nintedanib binds competitively to the ATP binding pocket of these kinases and blocks the intracellular signaling cascades, which have been demonstrated to be involved in the pathogenesis of fibrotic tissue remodeling in interstitial lung diseases.
Pharmacodynamic effects: NSCLC: Tumour angiogenesis is an essential feature contributing to tumour growth, progression and metastasis formation and is predominantly triggered by the release of pro-angiogenic factors secreted by the tumour cell (i.e. VEGF and bFGF) to attract host endothelial as well as perivascular cells to facilitate oxygen and nutrient supply through the host vascular system. In preclinical disease models, nintedanib, as single agent, effectively interfered with the formation and maintenance of the tumour vascular system resulting in tumour growth inhibition and tumour stasis. In particular, treatment of tumour xenografts with nintedanib led to a rapid reduction in tumour micro vessel density, pericytes vessel coverage and tumour perfusion.
Dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) measurements showed an anti-angiogenic effect of nintedanib in humans. It was not clearly dose dependent, but most responses were seen at doses of ≥ 200 mg. Logistic regression revealed a statistically significant association of the anti-angiogenic effect to nintedanib exposure. DCE-MRI effects were seen 24-48 h after the first drug intake and were preserved or even increased after continuous treatment over several weeks. No correlation of the DCE-MRI response and subsequent clinically significant reduction in target lesion size was found, but DCE-MRI response was associated with disease stabilization.
IPF/chronic fibrosing ILDs with a progressive phenotype/SSc-ILD: In in vitro studies using human cells nintedanib has been shown to inhibit processes assumed to be involved in the initiation of the fibrotic pathogenesis, the release of pro-fibrotic mediators from peripheral blood monocytic cells and macrophage polarisation to alternatively activated macrophages. Nintedanib has been demonstrated to inhibit fundamental processes in organ fibrosis, proliferation and migration of fibroblasts and transformation to the active myofibroblast phenotype and secretion of extracellular matrix. In animal studies in multiple models of IPF, SSc/SSc-ILD, RA-ILD and other organ fibrosis, nintedanib has shown anti-inflammatory effects and anti-fibrotic effects in the lung, skin, heart, kidney, and liver. Nintedanib also exerted vascular activity. It reduced dermal microvascular endothelial cell apoptosis and attenuated pulmonary vascular remodelling by reducing the proliferation of vascular smooth muscle cells, the thickness of pulmonary vessel walls and percentage of occluded pulmonary vessels.
CLINICAL TRIALS: Non-Small Cell Lung Cancer (NSCLC): Efficacy in the pivotal phase III trial LUME-Lung 1: The efficacy and safety of OFEV was investigated in 1314 patients with locally advanced, metastatic or recurrent NSCLC after one prior line of chemotherapy. 'Locally recurrent' was defined as local re-occurrence of the tumour without metastases at study entry. The trial including 658 patients (50.1%) with adenocarcinoma, 555 patients (42.2%) with squamous cell carcinoma, and 101 patients (7.7%) with other tumour histologies.
Patients were randomised (1:1) to receive nintedanib 200 mg orally twice daily in combination with 75 mg/m2 of i.v. docetaxel every 21 days (n = 655) or placebo orally twice daily in combination with 75 mg/m2 of docetaxel every 21 days (n = 659). Randomization was stratified according to Eastern Cooperative Oncology Group (ECOG) status (0 vs. 1), bevacizumab pretreatment (yes vs. no), brain metastasis (yes vs. no) and tumour histology (squamous vs. non-squamous tumour histology). Patient characteristics were balanced between treatment arms within the overall population and within the adenocarcinoma patients. In the overall population, 72.7% of the patients were male. The majority of patients were non-Asian (81.6%), the median age was 60.0 years, the baseline ECOG performance status was 0 (28.6%) or 1 (71.3%); one patient had a baseline ECOG performance status of 2. 5.8% of the patients had stable brain metastasis at study entry and 3.8% had prior bevacizumab treatment.
The disease stage was determined at the time of diagnosis using Union Internationale Contre le Cancer (UICC)/American Joint Committee on Cancer (AJCC) Edition 6 or Edition 7. In the overall population, 16.0% of the patients had disease stage < IIIB/IV, 22.4%, had disease stage IIIB and 61.6% had disease stage IV. 9.2% of the patients entered the study with locally recurrent disease stage as had been evaluated at baseline. For patients with tumour of adenocarcinoma histology, 15.8% had disease stage < IIIB/IV, 15.2%, had disease stage IIIB and 69.0% had disease stage IV.
5.8% of the adenocarcinoma patients entered the study with locally recurrent disease stage as had been evaluated at baseline.
The primary endpoint was progression-free survival (PFS) as assessed by an independent review committee (IRC). Overall survival (OS) was the key secondary endpoint. Other efficacy outcomes included objective response, disease control, change in tumour size and health-related quality of life.
As shown in Table 1, the addition of nintedanib to docetaxel led to a statistically significant reduction in the risk of progression or death by 21% for the overall population (HR 0.79; 95% CI: 0.68 - 0.92; p = 0.0019) as determined by the IRC. This result was confirmed in the follow-up PFS analysis (HR 0.85, 95% CI: 0.75 - 0.96; p = 0.0070) which included all events collected at the time of the final OS analysis. Overall survival analysis in the overall population did not reach statistical significance (HR 0.94; 95% CI: 0.83 - 1.05). Of note, pre-planned analyses according to histology showed statistically significant difference in OS between treatment arms in the adenocarcinoma population only. The addition of nintedanib to docetaxel led to a statistically significant reduction in the risk of progression or death by 23% for the adenocarcinoma population (HR 0.77; 95% CI: 0.62 - 0.96). In line with these observations, related study endpoints such as disease control and change in tumour size showed significant improvements. (See Table 1.)
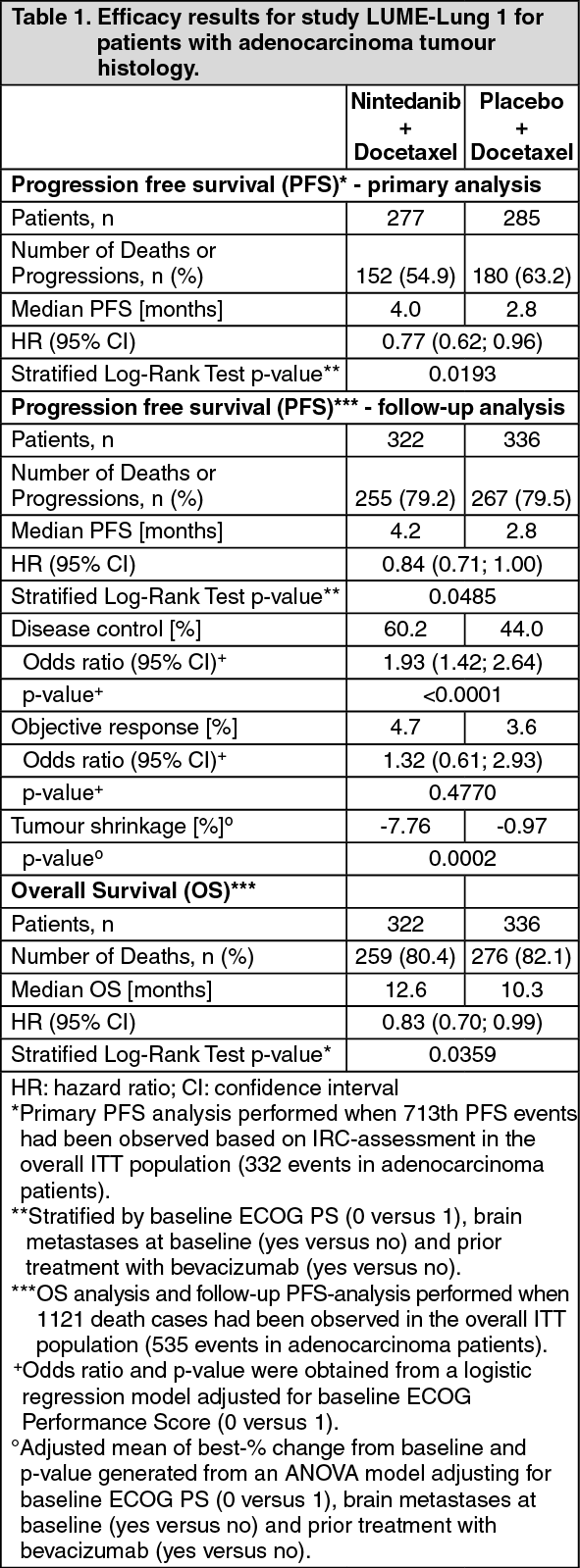 Click on icon to see table/diagram/image
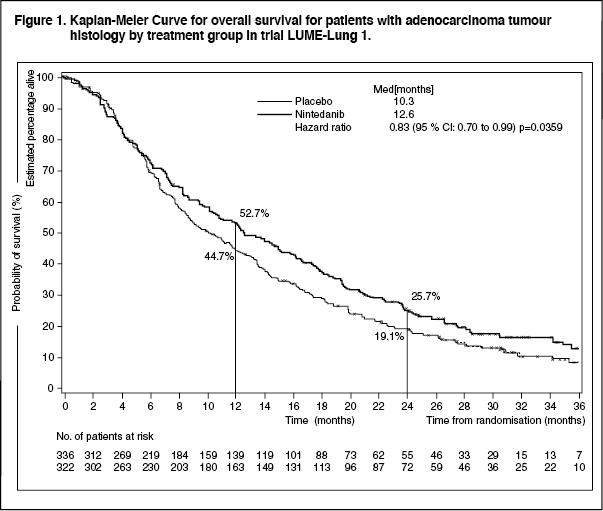
Click on icon to see table/diagram/imageA statistically significant improvement in OS favouring treatment with nintedanib plus docetaxel was demonstrated in patients with adenocarcinoma with a 17% reduction in the risk of death (HR 0.83, p = 0.0359) and a median OS improvement of 2.3 months (10.3 vs. 12.6 months, see Figure 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA pre specified evaluation was performed in the population of adenocarcinoma patients considered to have entered the study with a particularly poor treatment prognosis, namely, patients who progressed during or shortly after 1st line therapy prior to study entry. This population included those adenocarcinoma patients identified at baseline as having progressed and entered the study less than 9 months since start of their first-line therapy.
Treatment of these patients with nintedanib in combination with docetaxel reduced the risk of death by 25%, compared with placebo plus docetaxel (HR 0.75; 95% CI: 0.60 - 0.92; p = 0.0073). Median OS improved by 3 months (nintedanib: 10.9 months; placebo: 7.9 months).
In a post-hoc analysis in adenocarcinoma patients having progressed and entered the study ≥ 9 months since start of their first-line therapy the difference did not reach statistical significance (HR for OS: 0.89, 95% CI 0.66 - 1.19).
The proportion of adenocarcinoma patients with stage < IIIB/IV at diagnosis was small and balanced across treatment arms (placebo: 54 patients (16.1%); nintedanib: 50 patients, (15.5%)). The HR for these patients for PFS and OS was 1.24 (95% CI: 0.68, 2.28) and 1.09 (95% CI: 0.70, 1.70), respectively. However, the sample size was small, there was no significant interaction and the CI was wide and included the HR for OS of the overall adenocarcinoma population.
Idiopathic Pulmonary Fibrosis (IPF): The clinical efficacy of OFEV has been studied in patients with IPF in two phase III, randomised, double-blind, placebo-controlled studies with identical design (INPULSIS-1 and INPULSIS-2). Patients with FVC baseline < 50% predicted or carbon monoxide diffusing capacity (DLCO, corrected for haemoglobin) < 30% predicted at baseline were excluded from the trials.
Patients were randomized in a 3:2 ratio to treatment with OFEV 150 mg or placebo twice daily for 52 weeks.
The primary endpoint was the annual rate of decline in Forced Vital Capacity (FVC). The key secondary endpoints were change from baseline in Saint George's Respiratory Questionnaire (SGRQ) total score at 52 weeks and time to first acute IPF exacerbation.
Annual rate of decline in FVC: The annual rate of decline of FVC (in mL) was significantly reduced in patients receiving OFEV compared to patients receiving placebo. The treatment effect was consistent in both trials. See Table 2 for individual and pooled study results. (See Table 2 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image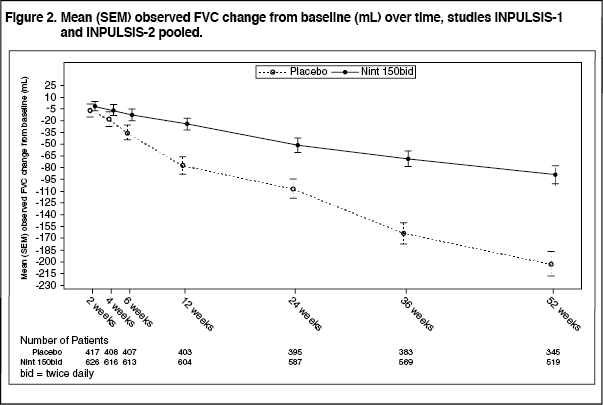 Click on icon to see table/diagram/image
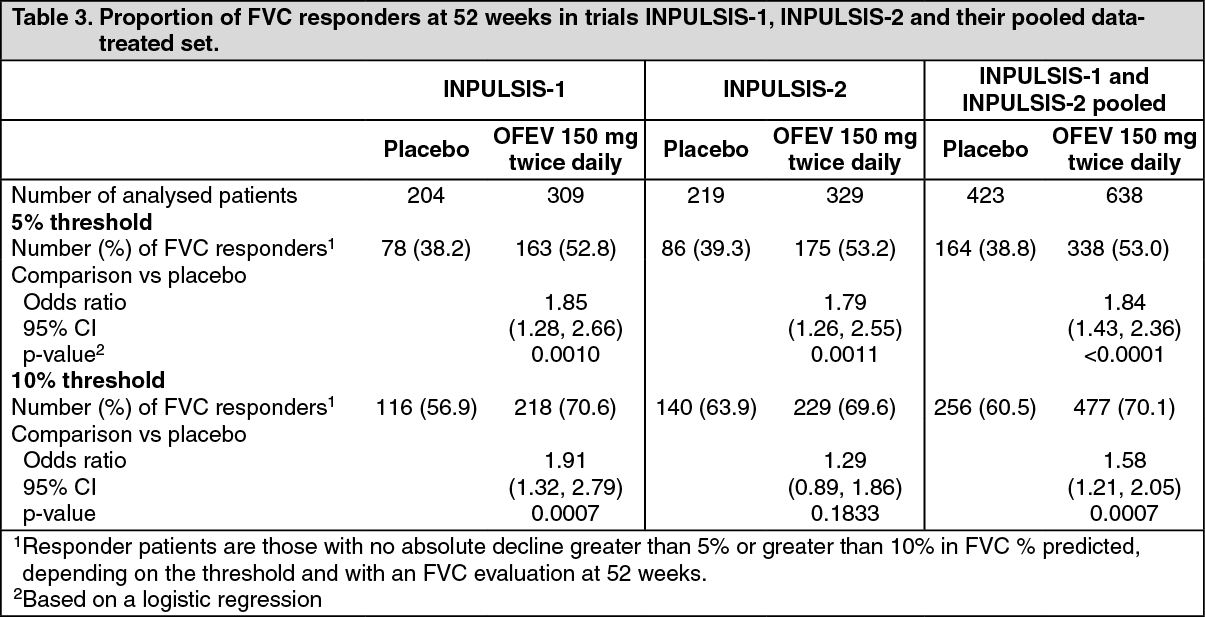
Click on icon to see table/diagram/imageFVC responder analysis: In both INPULSIS trials, the proportion of FVC responders, defined as patients with an absolute decline in FVC % predicted no greater than 5% (a threshold indicative of the increasing risk of mortality in IPF), was significantly higher in the OFEV group as compared to placebo. Similar results were observed in analyses using a conservative threshold of 10%. See Table 3 for individual and pooled study results. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTime to progression (≥ 10% absolute decline of FVC % predicted or death): In both INPULSIS trials, the risk of progression was statistically significantly reduced for patients treated with OFEV compared with placebo. In the pooled analysis, the HR was 0.60 indicating a 40% reduction in the risk of progression for patients treated with OFEV compared with placebo, (see Table 4).
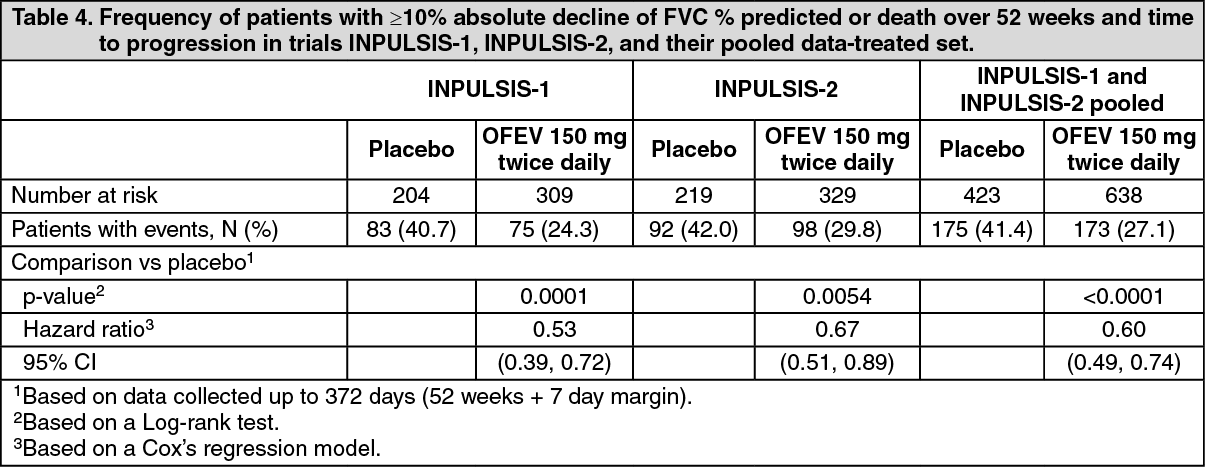 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageChange from baseline in SGRQ total score at week 52: St. George's Respiratory Questionnaire (SGRQ) total score measuring health related quality of life (HRQoL) was analysed at 52 weeks. In INPULSIS-2, patients receiving placebo had a larger increase from baseline SGRQ total score as compared to patients receiving OFEV 150 mg bid. The deterioration of HRQoL was smaller in the OFEV group; the difference between the treatment groups was statistically significant (-2.69; 95% CI: -4.95, -0.43; p=0.0197).
In INPULSIS-1, the increase from baseline in SGRQ total score at week 52 was comparable between OFEV and placebo (difference between treatment groups: -0.05; 95% CI: -2.50, 2.40; p=0.9657). In the pooled analysis of the INPULSIS trials, the estimated mean change from baseline to week 52 in SGRQ total score was smaller in the OFEV group (3.53) than in the placebo group (4.96), with a difference between the treatment groups of -1.43 (95% CI: -3.09, 0.23; p = 0.0923). Overall, the effect of OFEV on health-related quality of life as measured by the SGRQ total score is modest, indicating less worsening compared to placebo.
Time to first acute IPF exacerbation: In the INPULSIS-2 trial, the risk of first acute IPF exacerbation over 52 weeks was significantly reduced in patients receiving OFEV compared to placebo, in the INPULSIS-1 trial there was no difference between the treatment groups. In the pooled analysis of the INPULSIS trials, a numerically lower risk of first acute exacerbation was observed in patients receiving OFEV compared to placebo. See Table 5 for individual and pooled study results. (See Table 5.)
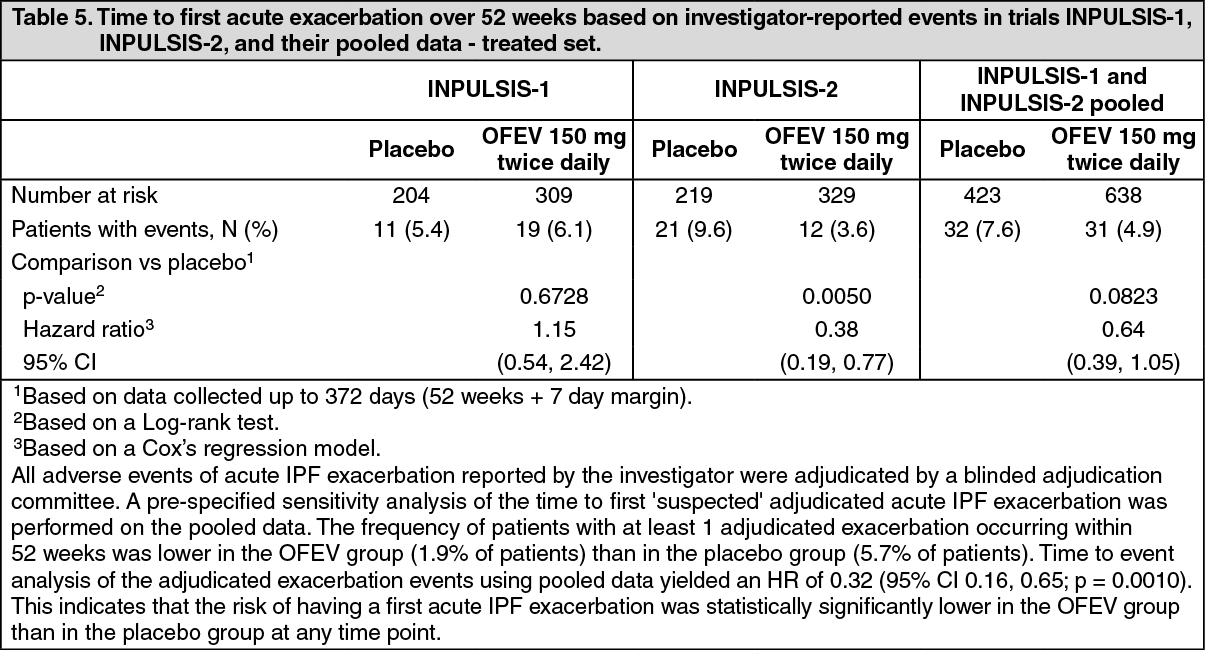 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSurvival analysis: In the pre-specified pooled analysis of survival data of the INPULSIS trials, overall mortality over 52 weeks was lower in the OFEV group (5.5%) compared with the placebo group (7.8%). The analysis of time to death resulted in a HR of 0.70 (95% CI 0.43, 1.12; p =0.1399). The results of all survival endpoints (such as on-treatment mortality and respiratory mortality) showed a consistent numerical difference in favour of OFEV (see Table 6).
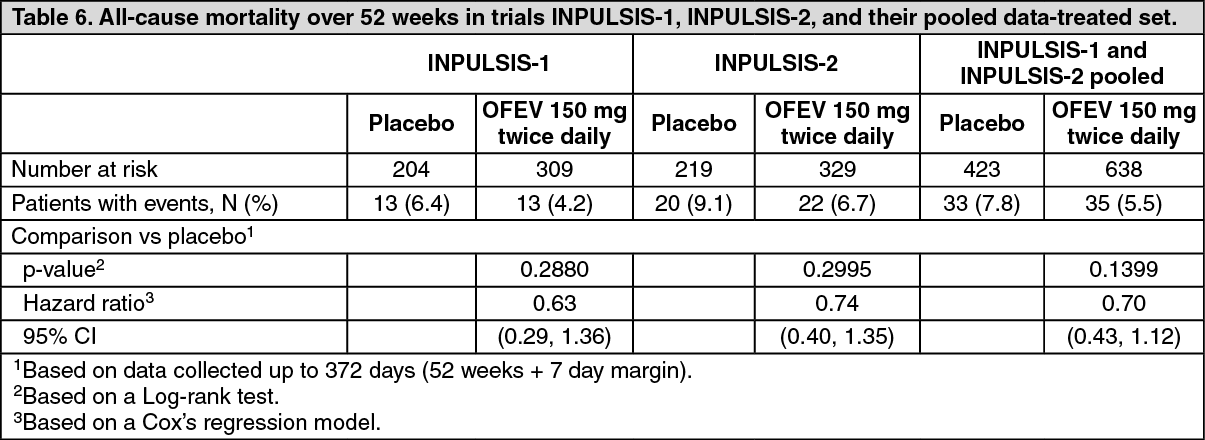 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSupportive evidence from the phase II trial (1199.30) OFEV 150 mg twice daily results: Additional evidence of efficacy is provided by the randomised, double-blind, placebo-controlled, dose finding phase II trial including a OFEV 150 mg bid dose group.
The primary endpoint, rate of decline in FVC over 52 weeks was lower in the OFEV arm (-0.060 L/year, N=84) than the placebo arm (-0.190 L/year, N=83). The estimated difference between the treatment groups was 0.131 L/year (95% CI 0.027, 0.235). The difference between the treatment groups reached nominal statistical significance (p = 0.0136).
The estimated mean change from baseline in SGRQ total score at 52 weeks was 5.46 for placebo, indicating worsening of the health-related quality of life and -0.66 for OFEV, indicating stable health-related quality of life. The estimated mean difference for OFEV compared with placebo was -6.12 (95% CI: -10.57, -1.67; p = 0.0071).
The number of patients with acute IPF exacerbations over 52 weeks was lower in the OFEV group (2.3%, N=86) compared to placebo (13.8%, N=87). The estimated hazard ratio of OFEV versus placebo was 0.16 (95% CI 0.04, 0.71; p = 0.0054).
Long-term treatment with OFEV in patients with IPF (INPULSIS-ON): An open-label extension trial of OFEV included 734 patients with IPF. Some patients were treated with OFEV for more than 5 years. Patients who completed the 52-week treatment period in an INPULSIS trial received open-label OFEV treatment in the extension trial INPULSIS-ON. Median exposure time for patients treated with OFEV in both the INPULSIS and INPULSIS-ON trials was 44.7 months (range 11.9-68.3). The adjusted annual rate of decline in FVC over 192 weeks was -135.1 (5.8) mL/year in all patients treated and were consistent with the annual rate of FVC decline in patients treated with OFEV in the INPULSIS phase III trials (-113.6 mL per year). The adverse event profile of OFEV in INPULSIS-ON was similar to that in the INPULSIS phase III trials.
Additional data from the phase IV INJOURNEY trial with OFEV 150 mg twice daily and add on pirfenidone: Concomitant treatment with OFEV and pirfenidone has been investigated in an exploratory open‐label, randomised trial of OFEV 150 mg twice daily with add‐on pirfenidone (titrated to 801 mg three times a day) compared to OFEV 150 mg twice daily alone in 105 randomised patients for 12 weeks. The primary endpoint was the percentage of patients with gastrointestinal adverse events from baseline to week 12. Gastrointestinal adverse events were frequent and in line with the established safety profile of each component. Diarrhoea, nausea and vomiting were the most frequent adverse events reported in 20 (37.7%) versus 16 (31.4%), in 22 (41.5%) versus 6 (11.8%) and in 15 (28.3%) versus 6 (11.8%) patients, treated with pirfenidone added to OFEV versus nintedanib alone, respectively.
Mean (SE) absolute changes from baseline in FVC at week 12 were -13.3 (17.4) mL in patients treated with nintedanib with add‐on pirfenidone (n=48) compared to -40.9 (31.4) mL in patients treated with nintedanib alone (n=44).
Chronic fibrosing Interstitial Lung Diseases (ILDs) with a progressive phenotype (PF-ILD): The clinical efficacy of OFEV has been studied in patients with chronic fibrosing ILDs with a progressive phenotype in a double-blind, randomised, placebo-controlled phase III trial (INBUILD). Patients with IPF were excluded. Patients with a clinical diagnosis of chronic fibrosing ILD were selected if they had relevant fibrosis (> 10% fibrotic features) on high resolution computed tomography (HRCT) and presented with clinical signs of progression. A total of 663 patients were randomised in a 1:1 ratio to receive either OFEV 150 mg bid or matching placebo for at least 52 weeks. (The median OFEV exposure over the whole trial was 17.4 months; and the mean OFEV exposure over the whole trial was 15.6 months). Randomisation was stratified based on HRCT fibrotic pattern as assessed by central readers. 412 patients with HRCT with usual interstitial pneumonia (UIP)-like fibrotic pattern and 251 patients with other HRCT fibrotic patterns were randomised. There were 2 co-primary populations defined for the analyses in this trial: all patients (the overall population) and patients with HRCT with UIP-like fibrotic pattern. Patients with other HRCT fibrotic patterns represented the 'complementary' population.
The primary endpoint was the annual rate of decline in Forced Vital Capacity (FVC) (in mL) over 52 weeks. Main secondary endpoints were absolute change from baseline in King's Brief Interstitial Lung Disease Questionnaire (K-BILD) total score at week 52, time to first acute ILD exacerbation or death over 52 weeks, and time to death over 52 weeks.
Patients had a mean (standard deviation [SD, Min-Max]) age of 65.8 (9.8, 27-87) years and a mean FVC percent predicted of 69.0% (15.6, 42-137). The underlying clinical ILD diagnoses in groups represented in the trial were hypersensitivity pneumonitis (26.1%), autoimmune ILDs (25.6%), idiopathic nonspecific interstitial pneumonia (18.9%), unclassifiable idiopathic interstitial pneumonia (17.2%), and other ILDs (12.2%).
Annual rate of decline in FVC: The annual rate of decline in FVC (in mL) over 52 weeks was significantly reduced by 107.0 mL in patients receiving OFEV compared to patients receiving placebo (Table 7) corresponding to a relative treatment effect of 57.0%. (See Table 7.)
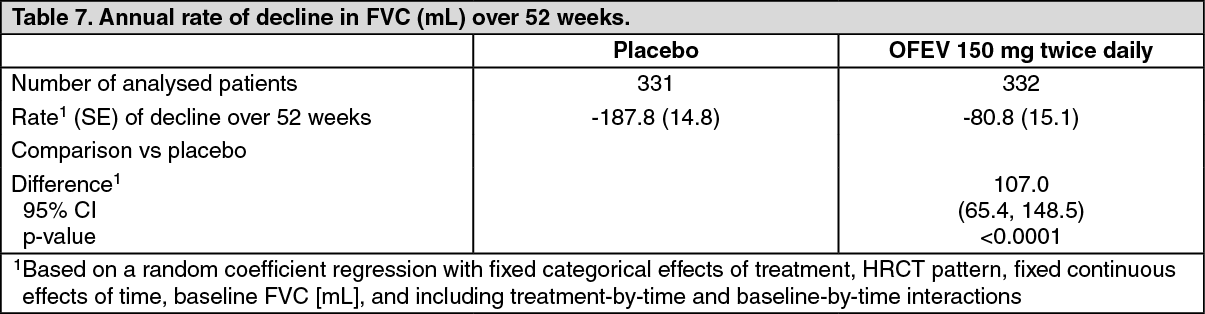 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSimilar results were observed in the co-primary population of patients with HRCT with UIP-like fibrotic pattern: the annual rate of decline in FVC was -211.1 mL/year in the placebo group (n=206) and -82.9 mL/year in the OFEV group (n=206). The difference between the treatment groups was 128.2 mL/year (95% CI: 70.8, 185.6; p<0.0001). Further, the treatment effect was consistent in the complementary population of patients with other HRCT fibrotic patterns. The annual rate of decline in FVC was -154.2 mL/year in the placebo group (n=125) and -79.0 mL/year in the OFEV group (n=126). The difference between the treatment groups was 75.2 mL/year (95% CI: 15.5, 135.0) with a nominal p-value < 0.05 (p=0.014). (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe robustness of the effect of OFEV in reducing the annual rate of decline in FVC was confirmed in all pre-specified sensitivity analyses and consistent results were observed in all pre-specified subgroups (e.g. gender, age group, race, baseline FVC % predicted, and original underlying clinical ILD diagnosis in groups).
Figure 4 shows the evolution of change in FVC from baseline over time in the treatment groups. (See Figure 4.)
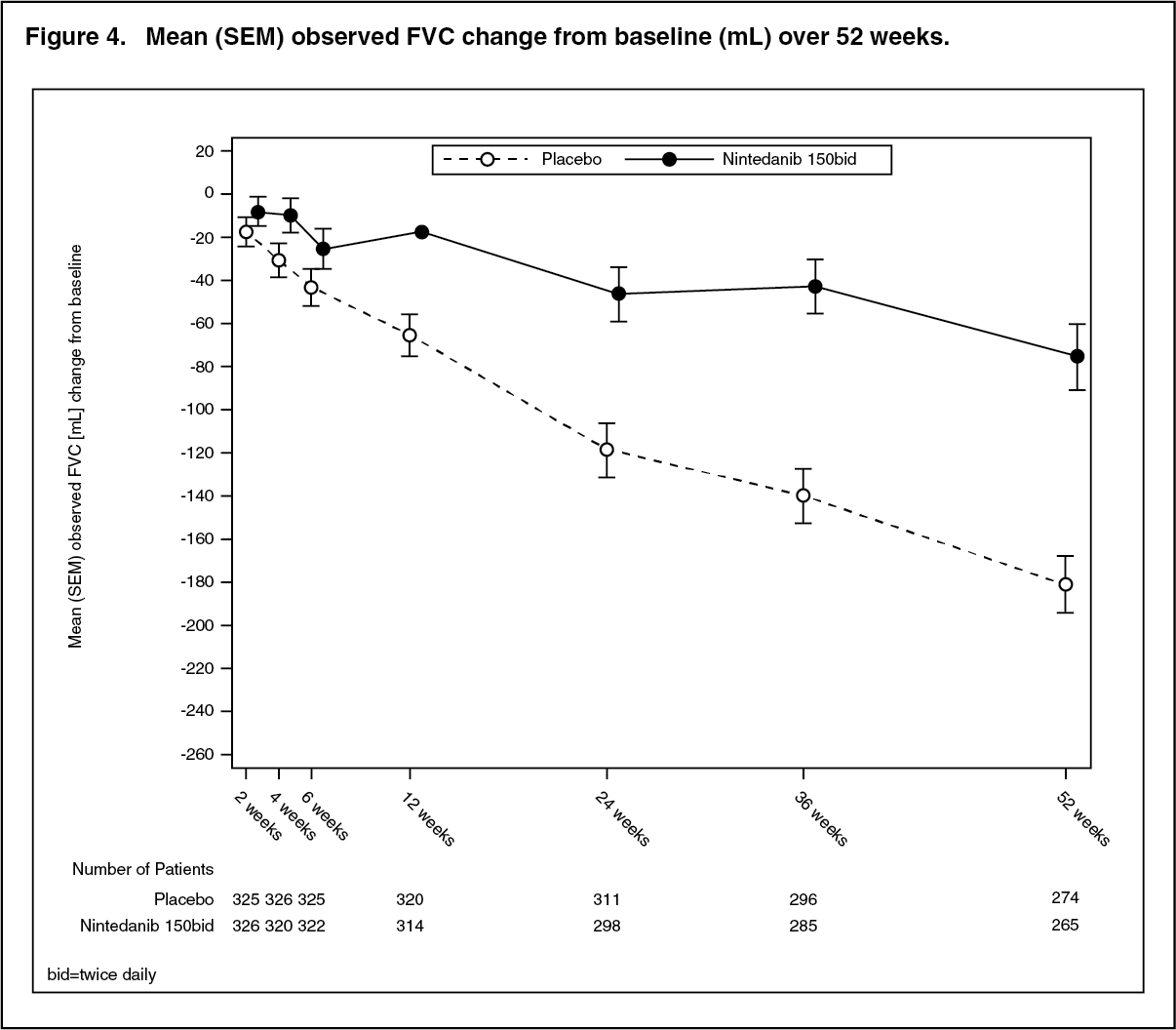 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn addition, favourable effects of OFEV were observed on the adjusted mean absolute change from baseline in FVC % predicted at week 52. The adjusted mean absolute change from baseline to week 52 in FVC % predicted was lower in the nintedanib group (-2.62%) than in the placebo group (-5.86%). The adjusted mean difference between the treatment groups was 3.24 (95% CI: 2.09, 4.40, nominal p<0.0001).
FVC responder analysis: The proportion of FVC responders, defined as patients with a relative decline in FVC % predicted no greater than 5%, was higher in the OFEV group as compared to placebo. Similar results were observed in analyses using a threshold of 10% (see Table 8).
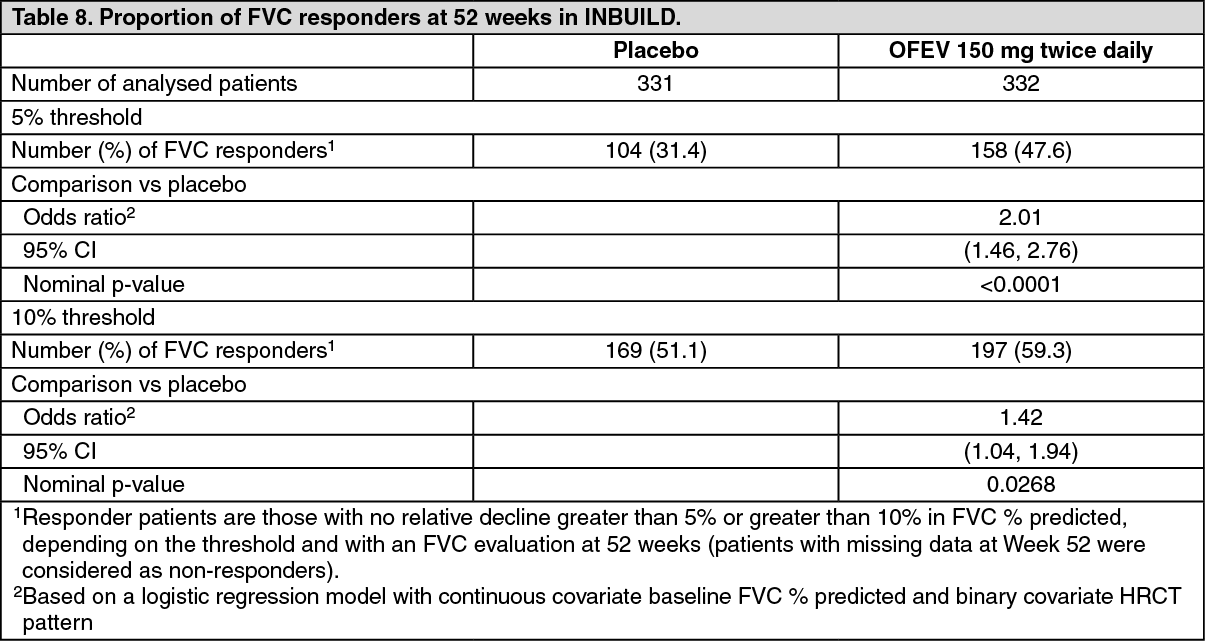 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTime to first acute ILD exacerbation or death: The proportion of patients with at least one event of first acute ILD exacerbation or death over 52 weeks was 7.8% in the OFEV group and 9.7% in the placebo group. The risk of having an event of first acute ILD exacerbation or death was numerically lower in the OFEV group compared to placebo: HR 0.80 (95% CI: 0.48, 1.34; nominal p=0.3948).
When analysing data over the whole trial, the risk of first acute ILD exacerbation or death further decreased in the OFEV group compared with the placebo group: the HR was 0.67 (95% CI: 0.46, 0.98; nominal p=0.0387), indicating a 33% reduction in the risk of first acute ILD exacerbation or death in patients receiving OFEV compared to placebo (see Figure 5).
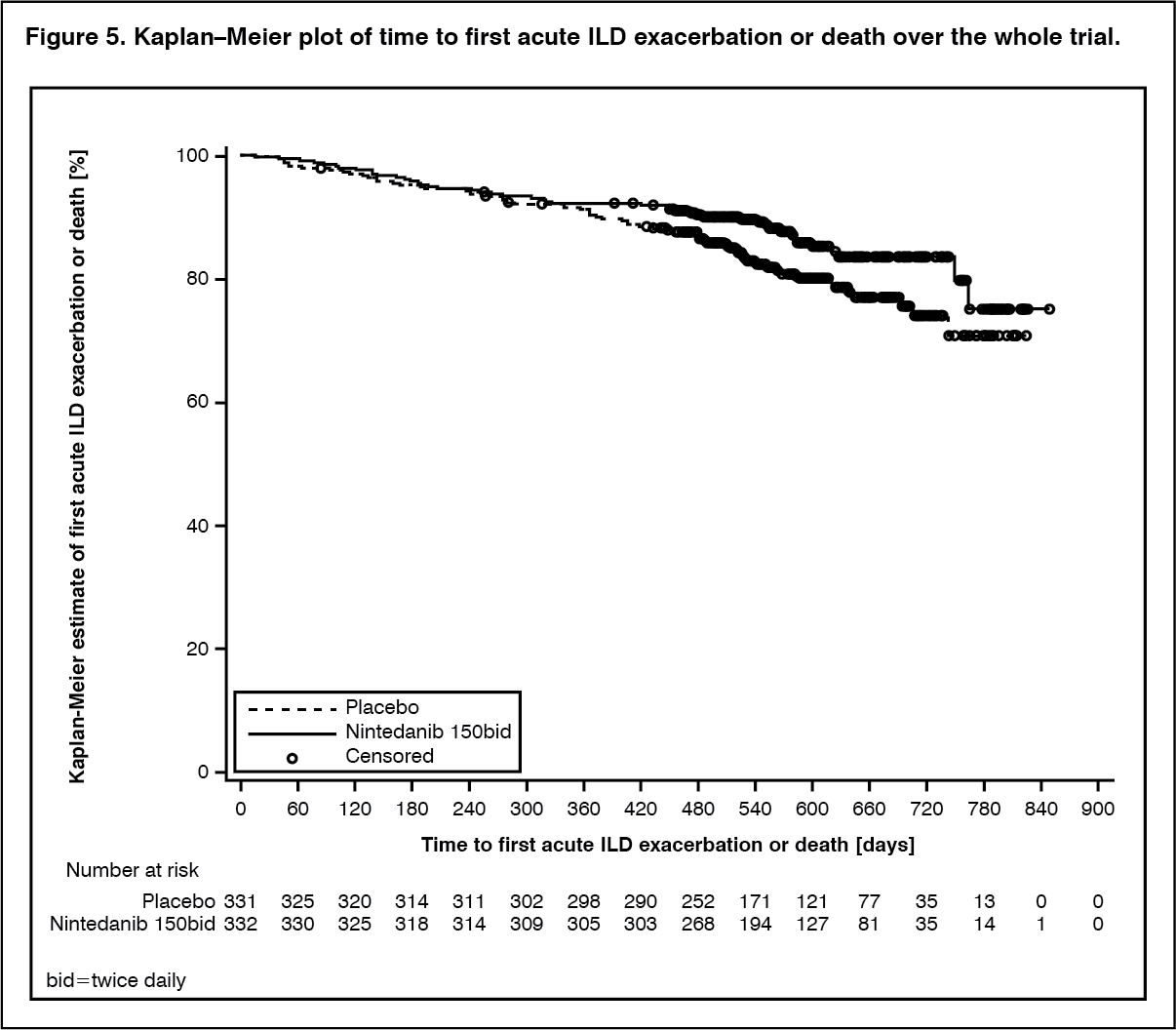 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSurvival analysis: The proportion of patients who died over 52 weeks was 4.8% in the OFEV group compared to 5.1% in the placebo group. The HR was 0.94 (95% CI: 0.47, 1.86; nominal p=0.8544).
In the analysis of data over the whole trial, the risk of death was lower in the OFEV group compared to the placebo group. The HR was 0.78 (95% CI: 0.50, 1.21; nominal p=0.2594), indicating a 22% reduction in the risk of death in patients receiving OFEV compared to placebo.
Time to progression (≥ 10% absolute decline of FVC % predicted) or death: In the INBUILD trial, the risk of progression (≥ 10% absolute decline of FVC % predicted) or death was reduced for patients treated with OFEV. The proportion of patients with an event over 52 weeks was 25.6% in the OFEV group and 37.5% in the placebo group. The HR was 0.65 (95% CI: 0.49, 0.85; nominal p=0.0017).
In the analysis of data over the whole trial, the proportion of patients with an event of progression (≥ 10% absolute decline of FVC % predicted) or death was 40.4% in the OFEV group and 54.7% in the placebo group. The HR was 0.66 (95% CI: 0.53, 0.83; p= 0.0003), indicating a 34% reduction in the risk of progression (≥ 10% absolute decline of FVC % predicted) or death in patients receiving OFEV compared to placebo.
Quality of life: In the INBUILD trial health related quality of life at 52 weeks was measured using the: Absolute change from baseline in King's Brief Interstitial Lung Disease Questionnaire (K-BILD) total score (range from 0-100, higher scores indicate a better health status).
Absolute change from baseline in Living with Pulmonary Fibrosis (L-PF) Symptoms dyspnoea domain score (range from 0-100, the higher the score the greater the impairment).
Absolute change from baseline in Living with Pulmonary Fibrosis (L-PF) Symptoms cough domain score (range from 0-100, the higher the score the greater the impairment).
The adjusted mean change from baseline in K-BILD total score at week 52 was -0.79 units in the placebo group and 0.55 in the OFEV group. The difference between the treatment groups was 1.34 (95% CI: -0.31, 2.98; nominal p=0.1115).
The adjusted mean absolute change from baseline in L-PF Symptoms dyspnoea domain score at week 52 was 4.28 in the OFEV group compared with 7.81 in the placebo group. The adjusted mean difference between the groups in favour of OFEV was -3.53 (95% CI: -6.14, -0.92; nominal p=0.0081). The adjusted mean absolute change from baseline in L-PF Symptoms cough domain score at week 52 was -1.84 in the OFEV group compared with 4.25 in the placebo group. The adjusted mean difference between the groups in favour of OFEV was -6.09 (95% CI: -9.65, -2.53; nominal p=0.0008).
Systemic Sclerosis associated Interstitial Lung Disease (SSc-ILD): The clinical efficacy of OFEV has been studied in in patients with SSc-ILD in a double-blind, randomised, placebo-controlled phase III trial (SENSCIS). Patients were diagnosed with SSc-ILD based upon the 2013 American College of Rheumatology/European League Against Rheumatism classification criteria for SSc and a chest high resolution computed tomography (HRCT) scan conducted within the previous 12 months. A total of 580 patients were randomised in a 1:1 ratio to receive either OFEV 150 mg bid or matching placebo for at least 52 weeks, of which 576 patients were treated. Randomisation was stratified by Antitopoisomerase Antibody status (ATA). Individual patients stayed on blinded trial treatment for up to 100 weeks (median OFEV exposure 15.4 months; mean OFEV exposure 14.5 months).
The primary endpoint was the annual rate of decline in Forced Vital Capacity (FVC) over 52 weeks. Key secondary endpoints were absolute change from baseline in the modified Rodnan Skin Score (mRSS) at week 52 and absolute change from baseline in the Saint George's Respiratory Questionnaire (SGRQ) total score at week 52.
In the overall population, 75.2% of the patients were female. The mean (standard deviation [SD, Min-Max]) age was 54.0 (12.2, 20-79) years. Overall, 51.9% of patients had diffuse cutaneous Systemic Sclerosis (SSc) and 48.1% had limited cutaneous SSc. The mean (SD) time since first onset of a non-Raynaud symptom was 3.49 (1.7) years. 49.0% of patients were on stable therapy with mycophenolate at baseline (46.5% mycophenolate mofetil, 1.9% mycophenolate sodium, 0.5% mycophenolic acid). The safety profile in patients with or without mycophenolate at baseline was comparable.
Annual rate of decline in FVC: The annual rate of decline of FVC (in mL) over 52 weeks was significantly reduced by 41.0 mL in patients receiving OFEV compared to patients receiving placebo (Table 9) corresponding to a relative treatment effect of 43.8%. (See Table 9.)
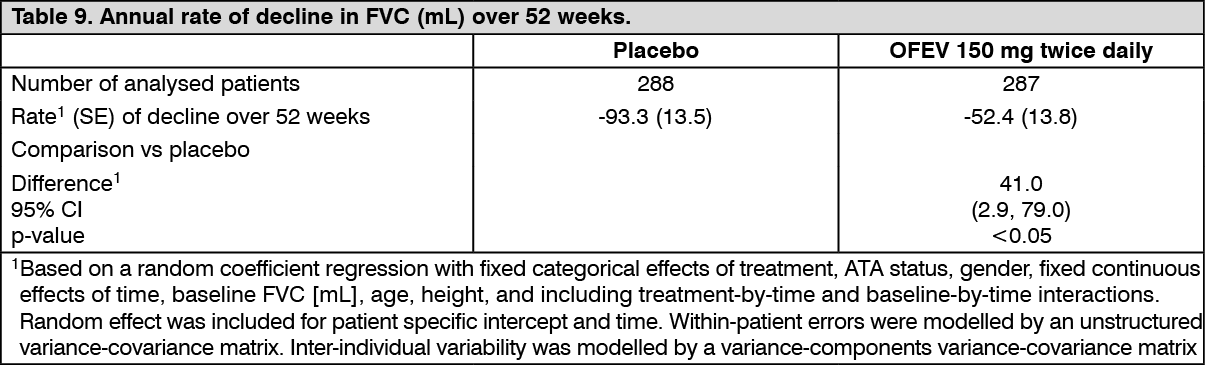 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe effect of OFEV in reducing the annual rate of decline in FVC was similar across pre-specified sensitivity analyses and no heterogeneity was detected in pre-specified subgroups (e.g. by age, gender, and mycophenolate use).
In addition, similar effects were observed on other lung function endpoints, e.g absolute change from baseline in FVC in mL at week 52 (Figure 6 and Table 10) and rate of decline in FVC in % predicted over 52 weeks (Table 11) providing further substantiation of the effects of OFEV on slowing progression of SSc-ILD. Furthermore, fewer patients in the OFEV group had an absolute FVC decline >5% predicted (20.6% in the OFEV group vs. 28.5% in the placebo group, OR=0.65, p=0.0287). The relative FVC decline in mL >10% was comparable between both groups (16.7% in the OFEV group vs. 18.1% in the placebo group, OR=0.91, p=0.6842). In these analyses, missing FVC values at week 52 were imputed with the patient's worst value on treatment.
An exploratory analysis of data up to 100 weeks (maximum treatment duration in SENSCIS) suggested that the on treatment effect of OFEV on slowing progression of SSc-ILD persisted beyond 52 weeks. (See Figure 6, Tables 10 and 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image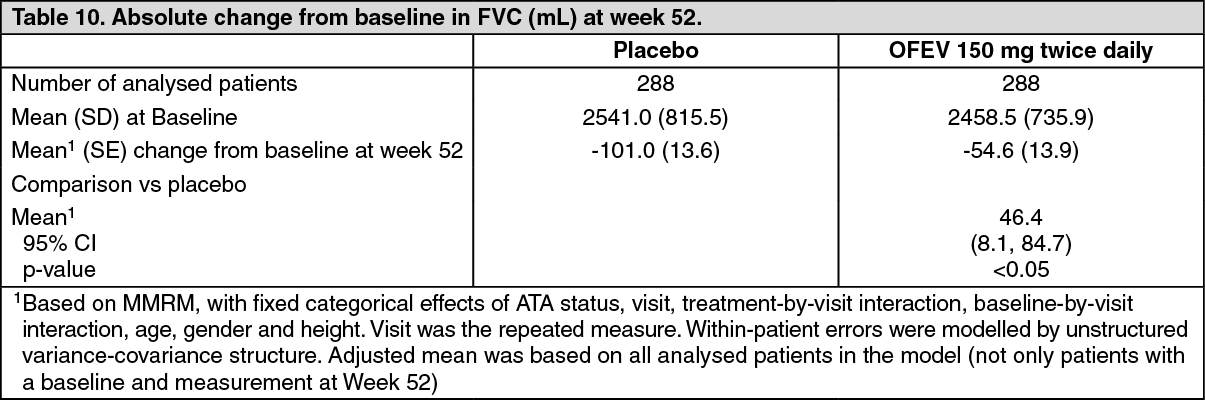 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image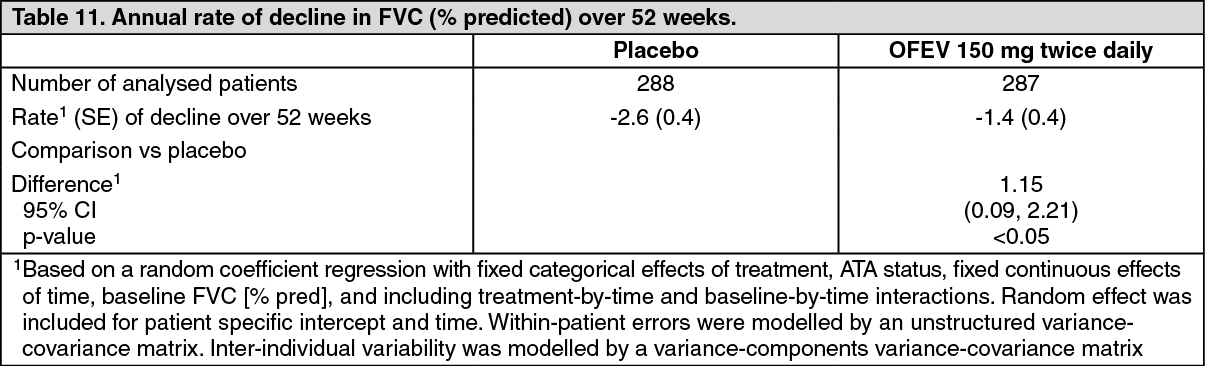 Click on icon to see table/diagram/image
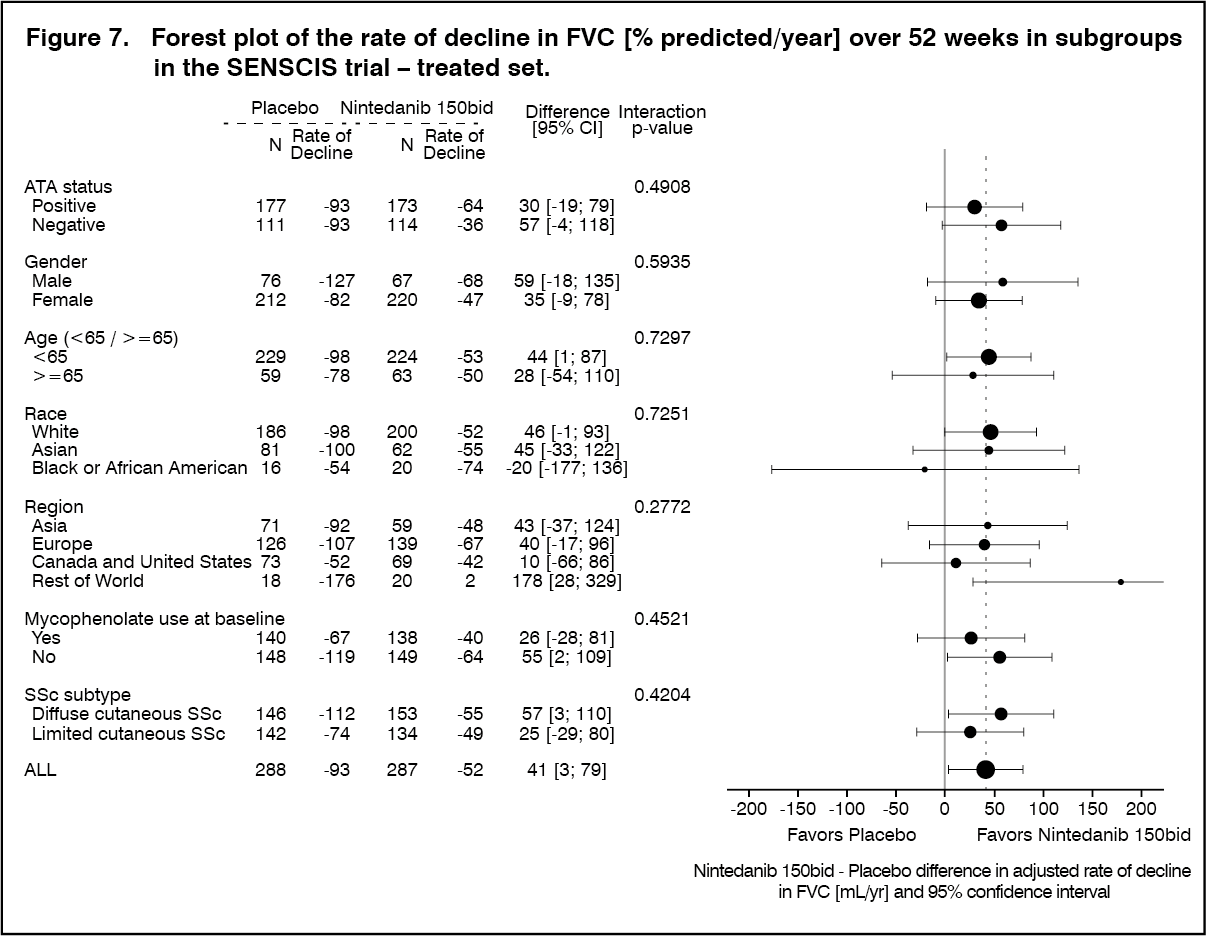
Click on icon to see table/diagram/imageThe annual rate of decline in FVC over 52 weeks was investigated in pre-specified subgroups by ATA status, gender, age, race, region, mycophenolate use at baseline, and SSc subtype. Overall, the analyses indicated a consistent treatment effect of nintedanib across all subgroups assessed. (See Figure 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageChange from baseline in Modified Rodnan Skin Score (mRSS) at week 52: The adjusted mean absolute change from baseline in mRSS at week 52 was comparable between the OFEV group (-2.17 (95% CI -2.69, -1.65)) and the placebo group (-1.96 (95% CI -2.48, -1.45)). The adjusted mean difference between the treatment groups was -0.21 (95% CI -0.94, 0.53; p = 0.5785).
Change from baseline in St. George's Respiratory Questionnaire (SGRQ) total score at week 52: The adjusted mean absolute change from baseline in SGRQ total score at week 52 was comparable between the OFEV group (0.81 (95% CI -0.92, 2.55)) and the placebo group (-0.88 (95% CI -2.58, 0.82)). The adjusted mean difference between the treatment groups was 1.69 (95% CI -0.73, 4.12; p = 0.1711).
Survival analysis: Mortality over the whole trial was comparable between the OFEV group (N = 10; 3.5%) and the placebo group (N = 9; 3.1%). The analysis of time to death over the whole trial resulted in a HR of 1.16 (95% CI 0.47, 2.84; p = 0.7535).
Quality of Life: NSCLC: Treatment with nintedanib did not significantly change the time to deterioration of the pre-specified symptoms cough, dyspnoea and pain, but resulted in a significant deterioration in the diarrhoea symptom scale. Nevertheless, the overall treatment benefit of nintedanib was observed without adversely affecting self-reported quality of life.
Effect on QT interval: QT/QTc measurements were recorded and analysed from a dedicated study comparing nintedanib monotherapy against sunitinib monotherapy in patients with renal cell carcinoma. In this study single oral doses of 200 mg nintedanib as well as multiple oral doses of 200 mg nintedanib administered twice daily for 15 days did not prolong the QTcF interval.
NSCLC: However, no thorough QT-trial of nintedanib administered in combination with docetaxel was conducted.
Paediatric population: The safety and efficacy of OFEV in children aged 0-18 years have not been established.
Pharmacokinetics: The pharmacokinetics (PK) of nintedanib can be considered linear with respect to time (i.e. single-dose data can be extrapolated to multiple-dose data). Accumulation upon multiple administrations was 1.04-fold for Cmax and 1.38-fold for AUCτ. Nintedanib trough concentrations remained stable for more than one year.
Absorption: Nintedanib reached maximum plasma concentrations approximately 2 - 4 hours after oral administration as soft gelatine capsule under fed conditions (range 0.5 - 8 hours). The absolute bioavailability of a 100 mg dose was 4.69% (90% CI: 3.615 - 6.078) in healthy volunteers.
Absorption and bioavailability are decreased by transporter effects and substantial first-pass metabolism.
Nintedanib exposure increased dose-proportionally in the dose range of 50 - 450 mg once daily and 150 - 300 mg twice daily. Steady state plasma concentrations were achieved within one week of dosing at the latest.
After food intake, nintedanib exposure increased by approximately 20% compared to administration under fasted conditions (CI: 95.3 - 152.5%) and absorption was delayed (median tmax fasted: 2.00 hours; fed: 3.98 hours).
In an in vitro study, mixing nintedanib capsules with a small amount of apple sauce or chocolate pudding for up to 15 minutes did not have any impact on the pharmaceutical quality. Therefore, taking the capsules with soft food would not be expected to alter the clinical effect when taken immediately.
Distribution: Nintedanib follows at least bi-phasic disposition kinetics. After intravenous infusion, a high volume of distribution during the terminal phase (Vss: 1050 L, 45.0% gCV) was observed.
The in vitro protein binding of nintedanib in human plasma was high, with a bound fraction of 97.8%. Serum albumin is considered to be the major binding protein. Nintedanib is preferentially distributed in plasma with a blood to plasma ratio of 0.869.
Biotransformation: The prevalent metabolic reaction for nintedanib is hydrolytic cleavage by esterases resulting in the free acid moiety BIBF 1202. BIBF 1202 is subsequently glucuronidated by UGT enzymes, namely UGT 1A1, UGT 1A7, UGT 1A8, and UGT 1A10 to BIBF 1202 glucuronide.
Only a minor extent of the biotransformation of nintedanib consisted of CYP pathways, with CYP 3A4 being the predominant enzyme involved. The major CYP-dependent metabolite could not be detected in plasma in the human ADME study. In vitro, CYP-dependent metabolism accounted for about 5% compared to about 25% ester cleavage.
NSCLC: In preclinical in vivo experiments, BIBF 1202 did not show efficacy despite its activity at target receptors of the substance.
Elimination: Total plasma clearance after intravenous infusion was high (CL: 1390 mL/min, 28.8% gCV). Urinary excretion of the unchanged active substance within 48 hours was about 0.05% of the dose (31.5% gCV) after oral and about 1.4% of the dose (24.2% gCV) after intravenous administration; the renal clearance was 20 mL/min (32.6% gCV). The major route of elimination of drug related radioactivity after oral administration of [14C] nintedanib was via faecal/biliary excretion (93.4% of dose, 2.61% gCV). The contribution of renal excretion to the total clearance was low (0.649% of dose, 26.3% gCV). The overall recovery was considered complete (above 90%) within 4 days after dosing. The terminal half-life of nintedanib was between 10 and 15 h (gCV % approximately 50%).
Exposure-response relationship: NSCLC: In exploratory pharmacokinetic (PK)-adverse event analyses, higher exposure to nintedanib tended to be associated with liver enzyme elevations, but not with gastrointestinal adverse events. PK-efficacy analyses were not performed for clinical endpoints. Logistic regression revealed a statistically significant association between nintedanib exposure and DCE-MRI response.
IPF/chronic fibrosing ILDs with a progressive phenotype/SSc-ILD: Exposure-response analyses of patient with IPF, other chronic fibrosing ILDs with a progressive phenotype, and SSc-ILD indicated an Emax like relationship between exposure and the annual rate of decline in FVC with an EC50 of around 3 ng/mL (relative standard error: around 55%). For comparison, median observed nintedanib trough concentrations for 150 mg bid OFEV were about 10 ng/mL.
With respect to safety, there seemed to be a weak relationship between nintedanib plasma exposure and ALT and/or AST elevations. Actual administered dose might be the better predictor for the risk of developing diarrhea of any intensity, even if plasma exposure as risk determining factor could not be ruled out (see Precautions).
Intrinsic and Extrinsic Factors; Special Populations: The PK properties of nintedanib were similar in healthy volunteers, patients with IPF, patients with other chronic fibrosing ILDs with a progressive phenotype, patients with SSc-ILD, and cancer patients. Based on results of Population PK (PopPK) analyses and descriptive investigations, exposure to nintedanib was not influenced by sex (body weight corrected), mild and moderate renal impairment (estimated by creatinine clearance), liver metastases, ECOG performance score, alcohol consumption, or P-gp genotype. Population PK analyses indicated moderate effects on exposure to nintedanib depending on age, body weight, and race (see as follows). Based on the high inter- individual variability of exposure observed in the clinical trials these effects are not considered clinically relevant (see Precautions).
Age: Exposure to nintedanib increased linearly with age. AUCτ,ss decreased by 16% for a 45-year old patient (5th percentile) and increased by 13% for a 76-year old patient (95th percentile) relative to a patient with the median age of 62 years. The age range covered by the analysis was 29 to 85 years; approximately 5% of the population was older than 75 years.
Based on a PopPK model, an increase in nintedanib exposure of approximately 20 - 25% was observed in patients ≥ 75 years compared with patients under 65 years.
Body weight: An inverse correlation between body weight and exposure to nintedanib was observed. AUCτ,ss increased by 25% for a 50 kg patient (5th percentile) and decreased by 19% for a 100 kg patient (95th percentile) relative to a patient with the median weight of 71.5 kg.
Race: The geometric mean exposure to nintedanib was 33% higher in Chinese, Taiwanese, and Indian patients while it was 22% lower in Koreans compared to Caucasians (body weight corrected). Data from Black individuals was very limited but in the same range as for Caucasians.
Hepatic impairment: In a dedicated single dose phase I study and compared to healthy subjects, exposure to nintedanib based on Cmax and AUC was 2.2-fold higher in volunteers with mild hepatic impairment (Child Pugh A; 90% CI 1.3 - 3.7 for Cmax and 1.2 - 3.8 for AUC, respectively). In volunteers with moderate hepatic impairment (Child Pugh B), exposure was 7.6-fold higher based on Cmax (90% CI 4.4 - 13.2) and 8.7-fold higher (90% CI 5.7 - 13.1) based on AUC, respectively, compared to healthy volunteers. Subjects with severe hepatic impairment (Child Pugh C) have not been studied.
Concomitant treatment with pirfenidone: IPF: In a dedicated pharmacokinetic study, concomitant treatment of OFEV with pirfenidone was investigated in patients with IPF. Group 1 received a single dose of 150 mg OFEV before and after uptitration to 801 mg pirfenidone three times a day at steady state. Group 2 received steady state treatment of 801 mg pirfenidone three times a day and had a PK profiling before and after at least 7 days of co‐treatment with 150 mg OFEV twice daily. In group 1, the adjusted geometric mean ratios (90% confidence interval (CI)) were 93% (57% ‐ 151%) and 96% (70% ‐ 131%) for Cmax and AUC0‐tz of nintedanib, respectively (n=12). In group 2, the adjusted geometric mean ratios (90% CI) were 97% (86% ‐ 110%) and 95% (86% ‐ 106%) for Cmax,ss and AUCτ,ss of pirfenidone, respectively (n=12). Based on these results, there is no evidence of a relevant pharmacokinetic drug‐drug interaction between nintedanib and pirfenidone when administered in combination.
Concomitant treatment with bosentan: In a dedicated pharmacokinetic study, concomitant treatment of OFEV with bosentan was investigated in healthy volunteers. Subjects received a single dose of 150 mg OFEV before and after multiple dosing of 125 mg bosentan twice daily at steady state. The adjusted geometric mean ratios (90% confidence interval (CI)) were 103% (86% - 124%) and 99% (91% - 107%) for Cmax and AUC0-tz of nintedanib, respectively (n=13), indicating that co-administration of nintedanib with bosentan did not alter the pharmacokinetics of nintedanib.
Concomitant treatment with oral hormonal contraceptives: In a dedicated pharmacokinetic study, female patients with SSc-ILD received a single dose of a combination of 30 µg ethinylestradiol and 150 µg levonorgestrel before and after twice daily dosing of 150 mg nintedanib for at least 10 days. The adjusted geometric mean ratios (90% confidence interval (CI)) were 117% (108% - 127%; Cmax) and 101% (93% - 111%; AUC0-tz) for ethinylestradiol and 101% (90% - 113%; Cmax) and 96% (91% - 102%; AUC0-tz) for levonorgestrel, respectively (n=15), indicating that co-administration of nintedanib has no relevant effect on the plasma exposure of ethinylestradiol and levonorgestrel.
Drug-Drug Interaction Potential: Metabolism: Drug-drug interactions between nintedanib and CYP substrates, CYP inhibitors, or CYP inducers are not expected, since nintedanib, BIBF 1202, and BIBF 1202 glucuronide did not inhibit or induce CYP enzymes preclinically nor was nintedanib metabolized by CYP enzymes to a relevant extent.
Transport: Nintedanib is a substrate of P-gp. For the interaction potential of nintedanib with this transporter, see Interactions. Nintedanib was shown to be not a substrate or inhibitor of OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 or MRP-2 in vitro. Nintedanib was also not a substrate of BCRP. Only a weak inhibitory potential on OCT-1, BCRP, and P-gp was observed in vitro which is considered to be of low clinical relevance. The same applies for nintedanib being a substrate of OCT-1.
Toxicology: General toxicology: Single dose toxicity studies in rats and mice indicated a low acute toxic potential of nintedanib. In repeat dose toxicology studies in rats, adverse effects (e.g. thickening of epiphyseal plates, lesions of the incisors) were mostly related to the mechanism of action (i.e. VEGFR-2 inhibition) of nintedanib. These changes are known from other VEGFR-2 inhibitors and can be considered class effects.
Diarrhoea and vomiting accompanied by reduced food consumption and loss of body weight were observed in toxicity studies in non-rodents.
There was no evidence of liver enzyme increases in rats, dogs, and Cynomolgus monkeys. Mild liver enzyme increases which were not due to serious adverse effects such as diarrhoea were only observed in Rhesus monkeys.
Reproduction toxicity: NSCLC: A study of male fertility and early embryonic development up to implantation in rats did not reveal effects on the male reproductive tract and male fertility.
In rats, embryo-foetal lethality and teratogenic effects were observed at exposure levels below human exposure at the maximum recommended human dose (MRHD) 200 mg twice daily. Effects on the development of the axial skeleton and on the development of the great arteries were also noted at subtherapeutic exposure levels.
In rabbits, embryo-foetal lethality and teratogenic effects comparable to those in rats were observed at an exposure slightly higher than in rats.
In rats, small amounts of radiolabelled nintedanib and/or its metabolites were excreted into the milk (≤ 0.5% of the administered dose).
From the 2-year carcinogenicity studies in mice and rats, there was no evidence for a carcinogenic potential of nintedanib.
Genotoxicity studies indicated no mutagenic potential for nintedanib.
IPF/chronic fibrosing ILDs with a progressive phenotype/SSc-ILD: A study of male fertility and early embryonic development up to implantation in rats did not reveal effects on the male reproductive tract and male fertility.
In rats, embryo-foetal lethality and teratogenic effects were observed at exposure levels below human exposure at the maximum recommended human dose (MRHD) of 150 mg twice daily. Effects on the development of the axial skeleton and on the development of the great arteries were also noted at subtherapeutic exposure levels.
In rabbits, embryo-foetal lethality and teratogenic effects comparable to those in rats were observed at an exposure slightly higher than in rats.
In rats, small amounts of radiolabelled nintedanib and/or its metabolites were excreted into the milk (≤ 0.5% of the administered dose).
From the 2-year carcinogenicity studies in mice and rats, there was no evidence for a carcinogenic potential of nintedanib.
Genotoxicity studies indicated no mutagenic potential for nintedanib.