


 Sign Out
Sign Out


For the management of selected adverse reactions, see Precautions. Information about selected adverse reactions observed from the LUME-Lung 1 trial is described as follows.
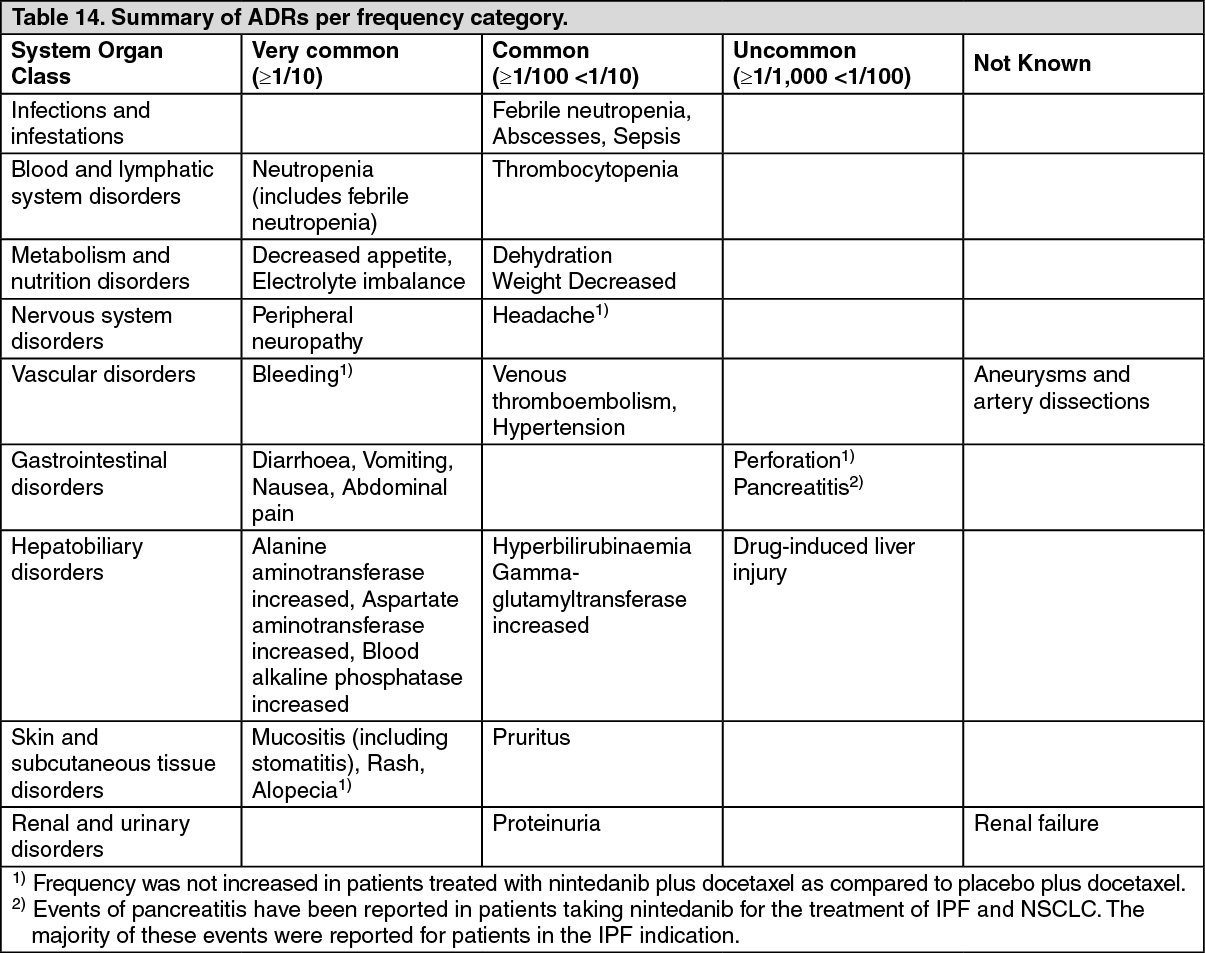
Tabulated list of adverse reactions Table 14 summarizes the frequencies of adverse drug reactions that were reported in the pivotal study LUME-Lung 1 for patients with NSCLC of adenocarcinoma tumour histology (n = 320).
The following terms are used to rank the ADRs by frequency: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from the available data).
Within each frequency grouping adverse reactions are presented in order of decreased seriousness. (See Table 14.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Diarrhoea: Diarrhoea occurred in 43.4% (≥ grade 3: 6.3%) of adenocarcinoma patients in the nintedanib arm. The majority of adverse reactions appeared in close temporal relationship with the administration of docetaxel. Most patients recovered from diarrhoea following treatment interruption, anti-diarrhoeal therapy and nintedanib dose reduction.
For recommended measures and dosing adjustments in case of diarrhoea, see Precautions and Dosage & Administration, respectively.
Liver enzyme elevations and hyperbilirubinaemia: Liver-related adverse reactions occurred in 42.8% of nintedanib-treated patients. Approximately one third of these patients had liver-related adverse reactions of ≥ grade 3 severity. In patients with increased liver parameters, the use of the established stepwise dose reduction scheme was the appropriate measure and discontinuation of treatment was only necessary in 2.2% of patients. In the majority of patients, elevations of liver parameters were reversible.
For information about special populations, recommended measures and dosing adjustments in case of liver enzyme and bilirubin elevations, see Precautions and Dosage & Administration, respectively.
Neutropenia, febrile neutropenia and sepsis: Sepsis and febrile neutropenia have been reported as subsequent complications of neutropenia. The rates of sepsis (1.3%) and febrile neutropenia (7.5%) were increased under treatment with nintedanib as compared to the placebo arm. It is important that the patient's blood counts are monitored during therapy, in particular during the combination treatment with docetaxel (see Precautions).
Bleeding: Although bleeding is an expected adverse reaction of nintedanib due to its mechanism of action, the bleeding incidence was comparable between the 2 treatment groups (placebo: 11.1%, nintedanib: 10.9%) in adenocarcinoma patients.
Perforation: As expected via its mechanism of action perforation might occur in patients treated with nintedanib. However, the frequency of patients with gastrointestinal perforation was low.
Peripheral neuropathy: Peripheral neuropathy is also known to occur with docetaxel treatment. Peripheral neuropathy was reported in 16.5% of patients in the placebo arm and in 19.1% of patients in the nintedanib arm.
IPF/chronic fibrosing ILDs with a progressive phenotype/SSc-ILD: Summary of the safety profile: Clinical trials in adult patients: OFEV has been studied in clinical trials including 1,529 patients suffering from Idiopathic Pulmonary Fibrosis (IPF), 663 patients with other chronic fibrosing Interstitial Lung Diseases (ILDs) with a progressive phenotype, and 576 patients with Systemic Sclerosis associated Interstitial Lung Disease (SSc-ILD).
The safety data provided in the following are based on: Two phase III, randomised, double-blind, placebo-controlled trials comparing treatment with OFEV 150 mg twice daily to placebo for 52 weeks (INPULSIS-1 and INPULSIS-2) in 1061 patients with IPF.
One phase III randomised, double-blind, placebo-controlled trial comparing treatment with OFEV 150 mg twice daily to placebo for at least 52 weeks in 663 patients with other chronic fibrosing ILDs with a progressive phenotype (INBUILD).
One phase III randomised, double-blind, placebo-controlled trial comparing treatment with OFEV 150 mg twice daily to placebo for at least 52 weeks in 576 patients with SSc-ILD (SENSCIS).
Data observed during the post‐marketing experience.
In clinical trials, the most frequently reported adverse reactions associated with the use of OFEV included diarrhoea, nausea and vomiting, abdominal pain, decreased appetite, weight decreased and hepatic enzyme increased.
The safety profile of OFEV in a long term extension trial in patients with IPF, treated from 1 up to more than 5 years, was consistent with that observed in the phase III trials (see Pharmacology: Pharmacodynamics: Clinical Trials under Actions).
For the management of selected adverse reactions see Precautions.
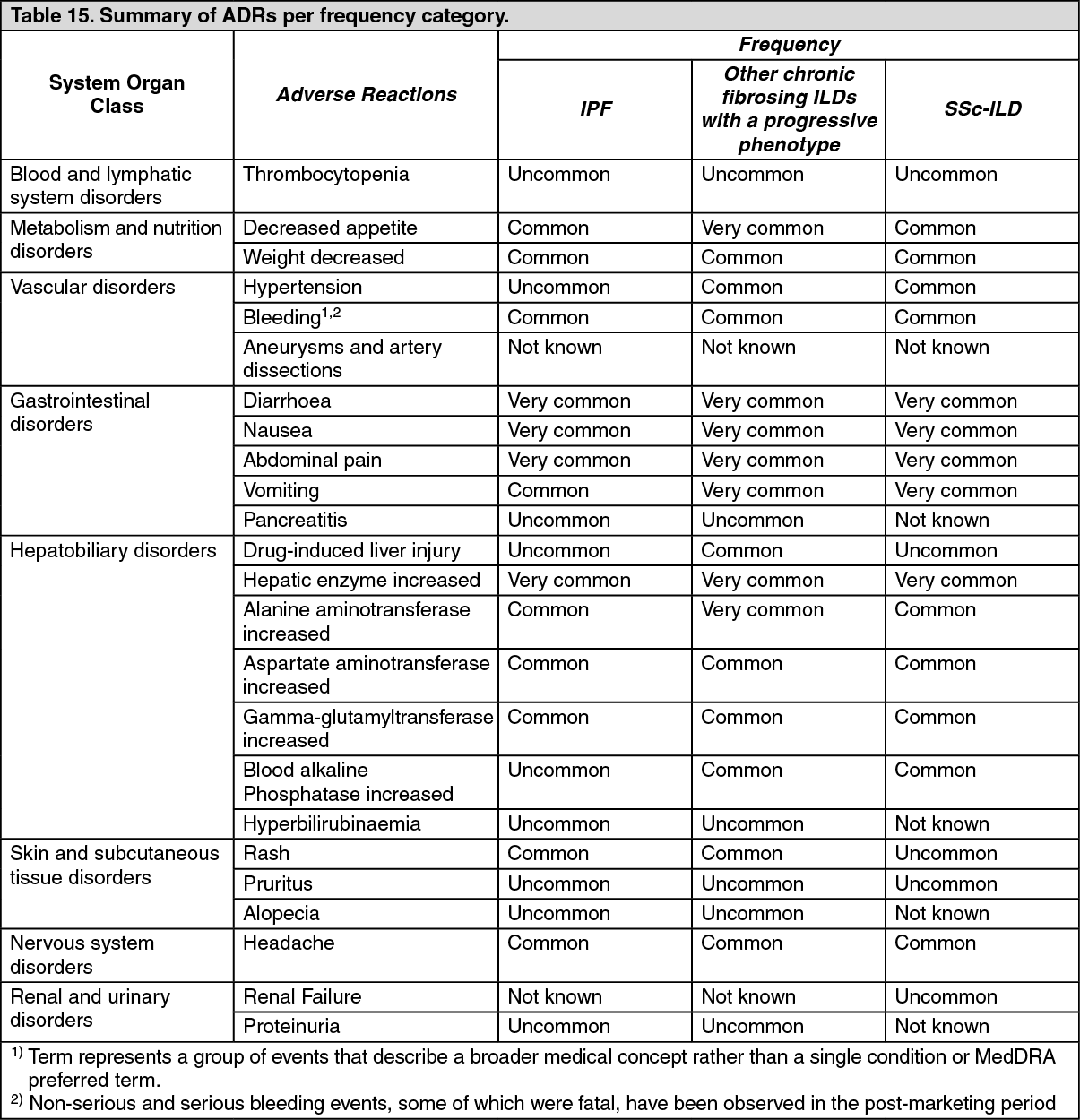
Tabulated list of adverse reactions: The table as follows provides a summary of the adverse reactions by MedDRA System Organ Class (SOC) and frequency category.
Frequency categories are defined using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from the available data).
Within each frequency grouping adverse reactions are presented in order of decreasing seriousness. (See Table 15.)
 Click on icon to see table/diagram/image
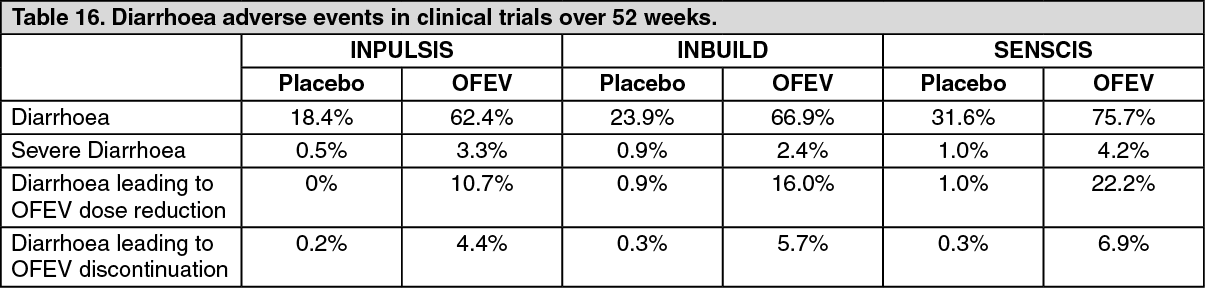
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Diarrhoea: In clinical trials (see Pharmacology: Pharmacodynamics under Actions), diarrhoea was the most frequent gastro-intestinal event reported. In most patients, the event was of mild to moderate intensity. More than two thirds of patients experiencing diarrhoea reported its first onset already during the first three months of treatment. In most patients, the events were managed by antidiarrhoeal therapy, dose reduction or treatment interruption (see Precautions). An overview of the reported diarrhoea events in the clinical trials is listed in Table 16. (See Table 16.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHepatic enzyme increased: In the INPULSIS trials, liver enzyme elevations (see Precautions) were reported in 13.6% versus 2.6% of patients treated with OFEV and placebo, respectively. In the INBUILD trial, liver enzyme elevations were reported in 22.6% versus 5.7% of patients treated with OFEV and placebo, respectively. In the SENSCIS trial, liver enzyme elevations were reported in 13.2% versus 3.1% of patients treated with OFEV and placebo, respectively. Elevations of liver enzymes were reversible and not associated with clinically manifest liver disease.
For further information about special populations, recommended measures and dosing adjustments in case of diarrhoea and hepatic enzyme increased, refer additionally to Precautions and Dosage & Administration, respectively.
View ADR Monitoring Form