Pharmacotherapeutic group: Tumor Necrosis Factor-Alpha (TNF-α) Inhibitors.
ATC code: L04AB02.
Pharmacology: Pharmacodynamics: Mechanism of Action: Infliximab products neutralize the biological activity of TNFα by binding with high affinity to the soluble and transmembrane forms of TNFα and inhibits binding of TNFα with its receptors. Infliximab does not neutralize TNFβ (lymphotoxin-α), a related cytokine that utilizes the same receptors as TNFα. Biological activities attributed to TNFα include: induction of pro-inflammatory cytokines such as interleukins (IL) 1 and 6, enhancement of leukocyte migration by increasing endothelial layer permeability and expression of adhesion molecules by endothelial cells and leukocytes, activation of neutrophil and eosinophil functional activity, induction of acute-phase reactants and other liver proteins, as well as tissue degrading enzymes produced by synoviocytes and/or chondrocytes. Cells expressing transmembrane TNFα bound by infliximab can be lysed
in vitro or
in vivo. Infliximab products inhibit the functional activity of TNFα in a wide variety of
in vitro bioassays utilizing human fibroblasts, endothelial cells, neutrophils, B and T lymphocytes and epithelial cells. The relationship of these biological response markers to the mechanism(s) by which infliximab products exert its clinical effects is unknown. Anti-TNFα antibodies reduce disease activity in the cotton-top tamarin colitis model, and decrease synovitis and joint erosions in a murine model of collagen-induced arthritis. Infliximab prevents disease in transgenic mice that develop polyarthritis as a result of constitutive expression of human TNFα, and when administered after disease onset, allows eroded joints to heal.
Pharmacodynamics: Elevated concentrations of TNFα have been found in involved tissues and fluids of patients with RA, Crohn's disease, UC, ankylosing spondylitis, psoriatic arthritis and plaque psoriasis. In RA, treatment with infliximab products reduced infiltration of inflammatory cells into inflamed areas of the joint as well as expression of molecules mediating cellular adhesion (E-selectin, intercellular adhesion molecule-1 [ICAM-1] and vascular cell adhesion molecule-1 [VCAM-1]), chemoattraction (IL-8 and monocyte chemotactic protein [MCP-1]) and tissue degradation (matrix metalloproteinase [MMP] 1 and 3). In Crohn's disease, treatment with infliximab products reduced infiltration of inflammatory cells and TNFα production in inflamed areas of the intestine, and reduced the proportion of mononuclear cells from the lamina propria able to express TNFα and interferon. After treatment with infliximab, patients with RA or Crohn's disease exhibited decreased levels of serum IL-6 and C-reactive protein (CRP) compared to baseline. Peripheral blood lymphocytes from infliximab product-treated patients showed no significant decrease in number or in proliferative responses to
in vitro mitogenic stimulation when compared to cells from untreated patients. In psoriatic arthritis, treatment with infliximab products resulted in a reduction in the number of T-cells and blood vessels in the synovium and psoriatic skin lesions as well as a reduction of macrophages in the synovium. In plaque psoriasis, infliximab products treatment may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which infliximab products exert their clinical effects is unknown.
Biosimilarity: In Study B5371002, serum hs-CRP was assessed as the PD biomarker and a component of the American College of Rheumatology (ACR) and disease activity score (DAS) assessments. Consistent with previous findings for TNFα inhibitors, mean serum hs-CRP concentrations decreased acutely in response to IXIFI and Remicade-EU treatments, and remained suppressed through Week 30. In the ITT population, mean changes from baseline (standard deviation) in hs-CRP were -12.2 (25.7) and -12.4 (30.0) mg/L at Week 30 for the IXIFI and Remicade-EU treatments, respectively, and were similar between the treatment arms over time.
CLINICAL STUDIES: Rheumatoid Arthritis: The efficacy of infliximab in adult patients with RA was assessed in two multicenter, randomized, double-blind, pivotal trials: ATTRACT and ASPIRE. In both studies concurrent use of stable doses of folic acid, oral corticosteroids (≤ 10 mg/day) and/or non-steroidal anti-inflammatory drugs were permitted.
The primary endpoints were the reduction of signs and symptoms as assessed by the American College of Rheumatology criteria (ACR20 for ATTRACT, landmark ACR-N for ASPIRE), the prevention of structural joint damage, and the improvement in physical function. A reduction in signs and symptoms was defined to be at least a 20% improvement (ACR20) in both tender and swollen joint counts, and in 3 of the following 5 criteria: (1) evaluator's global assessment, (2) patient's global assessment, (3) functional/disability measure, (4) visual analog pain scale and (5) erythrocyte sedimentation rate or C-reactive protein. ACR-N uses the same criteria as the ACR20, calculated by taking the lowest percent improvement in swollen joint count, tender joint count, and the median of the remaining 5 components of the ACR response. Structural joint damage (erosions and joint space narrowing) in both hands and feet was measured by the change from baseline in the total van der Heijde-modified Sharp score (0-440). The Health Assessment Questionnaire (HAQ; scale 0-3) was used to measure patients' average change from baseline scores over time, in physical function.
The ATTRACT trial evaluated responses at 30, 54 and 102 weeks in a placebo-controlled study of 428 patients with active rheumatoid arthritis despite treatment with methotrexate. Approximately 50% of patients were in functional Class III. Patients received placebo, 3 mg/kg or 10 mg/kg infliximab at weeks 0, 2 and 6, and then every 4 or 8 weeks thereafter. All patients were on stable methotrexate doses (median 15 mg/week) for 6 months prior to enrolment and were to remain on stable doses throughout the study.
Results from Week 54 (ACR20, HAQ and total van der Heijde-modified Sharp score) are shown in Table 1. Higher degrees of clinical response (ACR50 and ACR70) were observed in all infliximab groups at 30 and 54 weeks compared with methotrexate alone.
A reduction in the rate of the progression of structural joint damage (erosions and joint space narrowing) was observed in all infliximab groups at 54 weeks (Table 1).
The effects observed at 54 weeks were maintained through 102 weeks. Due to a number of treatment withdrawals, the magnitude of the effect difference between infliximab and the methotrexate alone group cannot be defined. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The ASPIRE trial evaluated responses at 54 weeks in 1004 methotrexate naïve patients with early (≤ 3 years disease duration, median 0.6 years) active rheumatoid arthritis (median swollen and tender joint count of 19 and 31, respectively). All patients received methotrexate (optimized to 20 mg/week by Week 8) and either placebo, 3 mg/kg or 6 mg/kg infliximab at weeks 0, 2, and 6 and every 8 weeks thereafter. Results from Week 54 are shown in Table 2.
After 54 weeks of treatment, both doses of infliximab and methotrexate resulted in statistically significantly greater improvement in signs and symptoms compared to methotrexate alone as measured by the proportion of patients achieving ACR20, 50 and 70 responses.
In ASPIRE, more than 90% of patients had at least two evaluable X-rays. Reduction in the rate of progression of structure damage was observed at weeks 30 and 54 in the infliximab and methotrexate groups compared to methotrexate alone. (See Table 2.)
Click on icon to see table/diagram/image
Adult Crohn's Disease: Induction Treatment in Moderately to Severely Active Crohn's Disease: The efficacy of a single dose treatment with infliximab was assessed in 108 adult patients with active Crohn's disease (Crohn's Disease Activity Index (CDAI) ≥ 220 ≤ 400) in a randomized, double-blinded, placebo-controlled, dose-response study. Of these 108 adult patients, 27 were treated with the recommended dosage of infliximab 5 mg/kg. All patients had experienced an inadequate response to prior conventional therapies. Concurrent use of stable doses of conventional therapies was permitted, and 92% of patients continued to receive these medications.
The primary endpoint was the proportion of patients who experienced a clinical response, defined as a decrease in CDAI by ≥ 70 points from baseline at the 4-week evaluation and without an increase in Crohn's disease medications or surgery for Crohn's disease. Patients who responded at Week 4 were followed to Week 12. Secondary endpoints included the proportion of patients in clinical remission at Week 4 (CDAI < 150) and clinical response over time.
At Week 4, following a single dose of study medication, 22/27 (81%) of infliximab-treated patients receiving a 5 mg/kg dose achieved a clinical response vs. 4/25 (16%) of the placebo-treated patients (p < 0.001). Also at Week 4, 13/27 (48%) of infliximab-treated patients achieved a clinical remission (CDAI < 150) vs. 1/25 (4%) of placebo-treated patients. A response was observed within 2 weeks, with a maximum response at 4 weeks. At the last observation at 12 weeks, 13/27 (48%) of infliximab-treated patients were still responding.
Maintenance Treatment in Moderately to Severely Active Crohn's Disease: The efficacy of repeated infusions with infliximab was studied in a 1-year clinical study. A total of 573 patients with active Crohn's disease (CDAI ≥ 220 ≤ 400) received a single infusion of 5 mg/kg at Week 0. Sixty-eight of these patients (12%) belonged to the population defined in the indication (see Indications/Uses). 335 patients (58%) responding to the 5 mg/kg infusion at Week 2 were randomized to one of three treatment groups; a placebo maintenance group, 5 mg/kg maintenance group and 10 mg/kg maintenance group, receiving repeated infusions at Week 2, 6 and every eight weeks.
At Week 30, a significantly greater proportion of patients in the combined infliximab maintenance treatment group (42%) achieved clinical remission, compared with patients in the placebo maintenance group (21%). Median time to loss of response was 46 weeks in the combined infliximab maintenance treatment group vs. 19 weeks in the placebo maintenance group (p < 0.001). Similar results were obtained in the subgroup analyses of the population defined in the indication (see Indications/Uses).
Improvements in quality of life measures were seen for both the IBDQ and SF-36 scores in the infliximab maintenance groups compared with the placebo maintenance group at Week 30 (p < 0.001).
Infliximab with or without AZA was assessed in a randomized, double-blind, active comparator study (SONIC) of 508 adult patients with moderate to severe Crohn's disease (CDAI ≥ 220 ≤ 450) who were naïve to biologics and immunosuppressants and had a median disease duration of 2.3 years. At baseline 27.4% of patients were receiving systemic corticosteroids, 14.2% of patients were receiving budesonide, and 54.3% of patients were receiving 5-ASA compounds. Patients were randomized to receive AZA monotherapy, infliximab monotherapy, or infliximab plus AZA combination therapy. Infliximab was administered at a dose of 5 mg/kg at weeks 0, 2, 6, and then every 8 weeks. AZA was given at a dose of 2.5 mg/kg daily.
The primary endpoint of the study was corticosteroid-free clinical remission at Week 26, defined as patients in clinical remission (CDAI of < 150) who, for at least 3 weeks, had not taken oral systemic corticosteroids (prednisone or equivalent) or budesonide at a dose > 6 mg/day. For results, see Table 3. The proportions of patients with mucosal healing at Week 26 were significantly greater in the infliximab plus AZA combination (43.9%, p < 0.001) and infliximab monotherapy groups (30.1%, p = 0.023) compared to the AZA monotherapy group (16.5%). (See Table 3.)
Click on icon to see table/diagram/image
Similar trends in the achievement of corticosteroid-free clinical remission were observed at Week 50. Furthermore, improved quality of life as measured by IBDQ was observed with infliximab.
Induction Treatment in Fistulizing Active Crohn's Disease: The efficacy was assessed in a randomized, double-blinded, placebo-controlled study in 94 adult patients with fistulizing Crohn's disease who had fistulae that were of at least 3 months' duration. Thirty-one of these patients were treated with infliximab 5 mg/kg. Approximately 93% of the patients had previously received antibiotic or immunosuppressive therapy.
Concurrent use of stable doses of conventional therapies was permitted, and 83% of patients continued to receive at least one of these medications. Patients received three doses of either placebo or infliximab at weeks 0, 2 and 6. Patients were followed up to 26 weeks. The primary endpoint was the proportion of patients who experienced a clinical response, defined as ≥ 50% reduction from baseline in the number of fistulae draining upon gentle compression on at least two consecutive visits (4 weeks apart), without an increase in medication for Crohn's disease.
68% (21/31) of infliximab-treated patients receiving a 5 mg/kg dose regimen achieved a clinical response vs. 26% (8/31) placebo-treated patients (p = 0.002). The median time to onset of response in the infliximab-treated group was 2 weeks. The median duration of response was 12 weeks. Additionally, closure of all fistulae was achieved in 55% of infliximab-treated patients compared with 13% of placebo-treated patients (p = 0.001).
Maintenance Treatment in Fistulizing Active Crohn's Disease: The efficacy of repeated infusions with infliximab in patients with fistulizing Crohn's disease was studied in a 1-year clinical study. A total of 306 patients received 3 doses of infliximab 5 mg/kg at Week 0, 2 and 6. At baseline, 87% of the patients had perianal fistulae, 14% had abdominal fistulae, 9% had rectovaginal fistulae. The median CDAI score was 180. At Week 14, 282 patients were assessed for clinical response and randomized to receive either placebo or 5 mg/kg infliximab every 8 weeks through Week 46.
Week-14 responders (195/282) were analyzed for the primary endpoint, which was time from randomization to loss of response (see Table 4). Corticosteroid tapering was permitted after Week 6. (See Table 4.)
Click on icon to see table/diagram/image
Beginning at Week 22, patients who initially responded to treatment and subsequently lost their response were eligible to cross over to active re-treatment every 8 weeks at a dose of infliximab 5 mg/kg higher than the dose to which they were originally randomized. Among patients in the infliximab 5 mg/kg group who crossed over because of loss of fistula response after Week 22, 57% (12/21) responded to re-treatment with infliximab 10 mg/kg every 8 weeks.
There was no significant difference between placebo and infliximab for the proportion of patients with sustained closure of all fistulas through Week 54, for symptoms such as proctalgia, abscesses and urinary tract infection or for number of newly developed fistulas during treatment.
Maintenance therapy with infliximab every 8 weeks significantly reduced disease-related hospitalizations and surgeries compared with placebo. Furthermore, a reduction in corticosteroid use and improvements in quality of life were observed.
Pediatric Crohn's Disease (6 to 17 years): In the REACH trial, 112 patients (6 to 17 years, median age 13.0 years) with moderate to severe, active Crohn's disease (median PCDAI of 40) and an inadequate response to conventional therapies were to receive 5 mg/kg infliximab at weeks 0, 2, and 6. All patients were required to be on a stable dose of 6-mercaptopurine (6-MP), azathioprine (AZA) or methotrexate (MTX) (35% were also receiving corticosteroids at baseline). Patients assessed by the investigator to be in clinical response at Week 10 were randomized and received 5 mg/kg infliximab either every 8-weeks or every 12-weeks as maintenance treatment regimen. If response was lost during treatment, crossing over to a higher dose (10 mg/kg) and/or shorter dosing interval (every 8-weeks) was allowed. 32 evaluable pediatric patients crossed over (9 subjects in the every 8-weeks and 23 subjects in the every 12-weeks maintenance groups). 24 of these patients (75.0%) regained clinical response after crossing over.
The proportion of subjects in clinical response at Week 10 was 88.4% (99/112). The proportion of subjects achieving clinical remission at Week 10 was 58.9% (66/112).
At Week 30, the proportion of subjects in clinical remission was higher in the every 8-week (59.6%, 31/52) than the every 12-week maintenance treatment group (35.3%, 18/51; p = 0.013) at Week 54, the figures were 55.8% (29/52) and 23.5% (12/51) in the every 8-weeks and every 12-weeks maintenance groups, respectively (p < 0.001).
Data about fistulas were derived from PCDAI scores. Of the 22 subjects that had fistulas at baseline, 63.6% (14/22), 59.1% (13/22) and 68.2% (15/22) were in complete fistula response at Week 10, 30 and 54, respectively, in the combined every 8-weeks and every 12-weeks maintenance groups.
In addition, statistically and clinically significant improvements in quality of life and height, as well as a significant reduction in corticosteroid use, were observed versus baseline.
Ulcerative Colitis: The safety and efficacy of infliximab was assessed in two (ACT 1 and ACT 2) randomized, double-blind, placebo-controlled clinical studies in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12; Endoscopy subscore ≥ 2) with an inadequate response to conventional therapies (oral corticosteroids, aminosalicylates and/or immunomodulators [6-MP, AZA]). Concomitant stable doses of oral aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted. In both studies, patients were randomized to receive either placebo, 5 mg/kg infliximab, or 10 mg/kg infliximab at weeks 0, 2, 6, 14 and 22, and in ACT 1 at weeks 30, 38 and 46. Corticosteroid taper was permitted after Week 8. (See Table 5.)
Click on icon to see table/diagram/image
The efficacy of infliximab through Week 54 was assessed in the ACT 1 trial.
At 54 weeks, 44.9% of patients in the combined infliximab treatment group were in clinical response compared to 19.8% in the placebo treatment group (p < 0.001). Clinical remission and mucosal healing occurred in greater proportion of patients in the combined infliximab treatment group compared to the placebo treatment group at Week 54 (34.5% vs. 16.5%, p < 0.001 and 46.1% vs. 18.2%, p < 0.001, respectively). The proportions of patients in sustained response and sustained remission at Week 54 were greater in the combined infliximab treatment group than in placebo treatment group (37.9% vs. 14.0%, p < 0.001; and 20.2% vs. 6.6%, p < 0.001, respectively).
Infliximab improved quality of life, confirmed by statistically and clinically significant improvement in both disease specific measure, IBDQ, and by improvement in the generic 36-item short form survey SF-36.
From baseline through Week 30 in the pooled data from ACT 1 and ACT 2, the mean number of hospitalizations was 50% lower in the combined infliximab treatment group than in the placebo treatment group (9 vs. 18 hospitalizations per 100 subjects, p = 0.005). No notable differences were observed between the 5 mg/kg and 10 mg/kg infliximab treatment groups.
A greater proportion of adult patients in the combined infliximab treatment group were able to discontinue corticosteroids in clinical remission compared to the placebo treatment group at both Week 30 (22.3% vs. 7.2%, p ≤ 0.001) and Week 54 (21.0% vs. 8.9%, p = 0.022).
Ulcerative Colitis in Pediatric Patients: The safety and efficacy of infliximab were assessed in a multicenter, randomized, open-label, parallel-group Phase 3 clinical study in 60 pediatric patients aged 6 through 17 years (median age 14.5 years) with moderately to severely active ulcerative colitis (Mayo score of 6 to 12; Endoscopic subscore ≥ 2) with an inadequate response to conventional therapies (Study Peds UC). At baseline 53% of patients were receiving immunomodulator therapy (6-mercaptopurine [6-MP]/ azathioprine [AZA]/ methotrexate [MTX]) and 62% of patients were receiving corticosteroids. Discontinuation of immunomodulators and corticosteroid taper were permitted after Week 0.
All patients received an induction regimen of 5 mg/kg infliximab at Weeks 0, 2, and 6. Patients (15) who did not respond to infliximab at Week 8 received no further drug and returned for safety follow-up. At Week 8, 45 patients were randomized in a 1:1 ratio to one of two maintenance treatment regimens: 5 mg/kg infliximab every 8 weeks (every 8-week) through Week 46 or every 12 weeks (every 12-week) through Week 42.
The primary endpoint was clinical response at Week 8, defined as a decrease from baseline in the Mayo score by ≥ 30% and ≥ 3 points, with a decrease in the rectal bleeding subscore of ≥ 1 or a rectal bleeding subscore of 0 or 1.
Major secondary endpoints included clinical remission measured by the Mayo score at Week 8, remission by the Pediatric Ulcerative Colitis Activity Index (PUCAI) score at Week 8 and Week 54, and mucosal healing at Week 8. For patients receiving corticosteroids at baseline, reduction in median corticosteroid use, and remission combined with elimination of corticosteroid use at Week 54 was evaluated.
Clinical Response, Clinical Remission and Mucosal Healing: Of the 60 patients treated, 44 (73.3%) were in clinical response at Week 8 (95% CI: 62.1%, 84.5%). The proportion of patients achieving clinical response at Week 8 was similar between those taking concomitant immunomodulators at baseline (72%) and those not taking concomitant immunomodulators at baseline (75%).
Clinical remission was defined by a Mayo score of ≤ 2 points, with no individual subscore > 1. Remission was also defined by a PUCAI score of < 10 points. At Week 8, infliximab induced clinical remission in 40% (24/60) of patients as measured by the Mayo score and in 33.3% (17/51) of patients as measured by the PUCAI score.
The proportion of patients in remission at Week 54 as measured by the PUCAI score was 38% (8/21) in the every 8-week maintenance treatment group and 18% (4/22) in the every 12-week maintenance treatment group.
Mucosal healing was defined as an endoscopy subscore (from the Mayo score) of 0 or 1. At Week 8, 68.3% (41/60) of patients were in mucosal healing with 33.3% (20/60) having an endoscopy subscore of 0 (indicating normal or inactive disease). Of the 9 patients who had an optional endoscopy at Week 54, 8 were in mucosal healing.
Overall, although some differences were noted between the age groups in the efficacy measures examined, efficacy was observed in both age groups and no consistent pattern indicating greater efficacy in one of the age groups was apparent. The differences between the 6 to 11 and the 12 to 17 year-old age groups, however, are difficult to assess because of the small sample sizes, particularly in the 6 to 11 year-old age group (15 patients).
Corticosteroid Use and Remission: Median average daily corticosteroid (prednisone equivalent) dose (mg/kg/day) through Week 54 is presented in Figure 1 for all randomized patients taking corticosteroids at baseline (14 patients in each maintenance treatment group). (See Figure 1.)
Click on icon to see table/diagram/image
At Week 54, the proportion of patients in clinical remission as measured by the PUCAI score was 38% (8/21) in the every 8-week maintenance group and 18% (4/22) in the every 12-week maintenance treatment group. For patients receiving corticosteroids at baseline, the proportion of patients in remission and not receiving corticosteroids at Week 54 was 38.5% (5/13) for the every 8-week and 0% (0/13) for the every 12-week maintenance treatment group.
For patients receiving corticosteroids at baseline in Study Peds UC, the proportion of these patients in remission and not receiving corticosteroids at Week 54 was 38.5% (5/13) for the every 8-week and 0% (0/13) for the every 12-week maintenance treatment group.
Ankylosing Spondylitis: Efficacy and safety were studied in a double-blind, placebo-controlled investigator-initiated, multicenter study evaluating infliximab in 70 patients with active ankylosing spondylitis (disease activity [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score > 4] and pain [NRS score > 4]). During the 3-month double-blind phase, patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6 (35 patients in each group). Starting at Week 12, placebo patients were switched to infliximab and all patients subsequently received 5 mg/kg infliximab every 6 weeks up to Week 54.
Treatment with infliximab resulted in improvement in signs and symptoms, as assessed by the BASDAI, with 57% of infliximab-treated patients achieving at least 50% reduction from baseline in BASDAI score (mean baseline score was 6.5 in the infliximab group and 6.3 in the placebo group, compared with 9% of placebo patients (p < 0.01). Improvement was observed at Week 2 and was maintained through Week 54. Physical function and quality of life (SF36) were improved similarly. In the trial, efficacy was not shown in HLA-B27 negative patients (n = 7).
Psoriatic Arthritis: Efficacy and safety were studied in a double-blind, placebo-controlled, multicenter study evaluating infliximab in 104 patients with active polyarticular psoriatic arthritis. In total 74 subjects were on at least one concomitant DMARD, and among those 58 patients were treated with methotrexate. During the 16-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6, and 14 (52 patients in each group). Starting at Week 16, placebo patients were switched to infliximab and all patients subsequently received 5 mg/kg infliximab every 8 weeks up to Week 46. After the first year of the study, 78 patients continued into an open-label extension to Week 98.
In the second trial (IMPACT 2), efficacy and safety of infliximab were studied in 200 patients with active psoriatic arthritis (≥ 5 swollen joints and ≥ 5 tender joints). 46% of patients continued on stable doses of methotrexate (≤ 25 mg/week). During the 24-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6, 14, and 22 (100 patients in each group). At Week 16, 47 placebo patients with < 10% improvement from baseline in both swollen and tender joint counts were switched to infliximab induction (early escape). At Week 24, all placebo-treated patients crossed over to infliximab induction. Dosing continued for all patients through Week 46.
Key efficacy results for IMPACT and IMPACT 2 are shown in Table 6 as follows: (See Table 6.)
Click on icon to see table/diagram/image
In IMPACT and IMPACT 2, clinical responses were observed as early as Week 2 and were maintained through Week 98 and Week 54 respectively. Efficacy has been demonstrated with or without concomitant use of methotrexate. Decreases in parameters of peripheral activity characteristic of psoriatic arthritis (such as number of swollen joints, number of painful/tender joints, dactylitis and presence of enthesopathy) were seen in the infliximab-treated patients.
Infliximab-treated patients demonstrated significant improvement in physical function as assessed by HAQ. Significant improvements in health-related quality of life were also demonstrated as measured by the physical and mental component summary scores of the SF-36 in IMPACT 2.
Psoriasis: The efficacy of infliximab was assessed in two multicenter, randomized, double-blind studies: SPIRIT and EXPRESS. Patients in both studies had plaque psoriasis (Body Surface Area [BSA] ≥ 10% and Psoriasis Area and Severity Index [PASI] score ≥ 12). The primary endpoint in both studies was the percent of patients who achieved ≥ 75% improvement in PASI from baseline at Week 10.
SPIRIT evaluated the efficacy of infliximab induction therapy in 249 patients with plaque psoriasis that had previously received PUVA or systemic therapy. Patients received either 3 or, 5 mg/kg infliximab or placebo infusions at weeks 0, 2 and 6. Patients with a PGA score ≥ 3 were eligible to receive an additional infusion of the same treatment at Week 26.
In SPIRIT, the proportion of patients achieving PASI 75 at Week 10 was 71.7% in the 3 mg/kg infliximab group, 87.9% in the 5 mg/kg infliximab group, and 5.9% in the placebo group (p < 0.001).
By Week 26, twenty weeks after the last induction dose, 30% of patients in the 5 mg/kg group and 13.8% of patients in the 3 mg/kg group were PASI 75 responders. Between weeks 6 and 26, symptoms of psoriasis gradually returned with a median time to disease relapse of > 20 weeks. No rebound was observed.
EXPRESS evaluated the efficacy of infliximab induction and maintenance therapy in 378 patients with plaque psoriasis. Patients received 5 mg/kg infliximab or placebo infusions at weeks 0, 2 and 6 followed by maintenance therapy every 8 weeks through Week 22 in the placebo group and through Week 46 in the infliximab group. At Week 24, the placebo group crossed over to infliximab induction therapy (5 mg/kg) followed by infliximab maintenance therapy (5 mg/kg). Nail psoriasis was assessed using the Nail Psoriasis Severity Index (NAPSI). Prior therapy with PUVA, methotrexate, cyclosporin, or acitretin had been received by 71.4% of patients, although they were not necessarily therapy resistant. Key results are presented in Table 7. In infliximab-treated subjects, significant PASI 50 responses were apparent at the first visit (Week 2) and PASI 75 responses by the second visit (Week 6). Efficacy was similar in the subgroup of patients that were exposed to previous systemic therapies compared to the overall study population. (See Table 7.)
Click on icon to see table/diagram/image
Significant improvements from baseline were demonstrated in DLQI (p < 0.001) and the physical and mental component scores of the SF 36 (p < 0.001 for each component comparison).
IXIFI Clinical Study - Rheumatoid Arthritis: The biosimilar clinical development program for IXIFI included a randomized, double-blind, active-controlled trial in subjects with moderately to severely active RA who have had an inadequate response to background MTX.
Study B5371002 was a multi-national, double-blind, randomized, comparative efficacy and safety study of 650 subjects designed to demonstrate the absence of clinically meaningful differences in the efficacy, safety and immunogenicity of IXIFI and the reference product (Remicade, sourced from the EU), and to evaluate the safety and immunogenicity of IXIFI after treatment transition from Remicade to IXIFI. The same dose regimen was followed for 3 mg/kg at Weeks 0, 2, and 6, followed by a maintenance regimen of every 8 weeks, with a one-time escalation to 5 mg/kg occurring on or after Week 14 for insufficient efficacy. The primary efficacy endpoint was ACR20 response rate at Week 14. Secondary endpoints included the ACR20 time course to Week 30, ACR50/70, DAS28-CRP, European League Against Rheumatism (EULAR) response, and ACR/EULAR remission evaluations. At Week 30, 50% of the Remicade arm were blindly re-randomized to the IXIFI arm. At Week 54, all patients received open label IXIFI for an additional 24 weeks.
For the ACR20 primary endpoint at Week 14, response rates were 62.7% for IXIFI and 64.1% for Remicade. In both ITT and PP populations, the 2-sided 95% CIs and 90% CIs of the treatment difference in Week 14 ACR20 response rate between the 2 groups were entirely contained within a symmetric equivalence margin of (-13.5% to 13.5%), and an asymmetric equivalence margin of (-12% to 15%), demonstrating therapeutic equivalence (similarity) between IXIFI and Remicade treatments.
Similar responses between IXIFI and Remicade treatments were observed at each study visit up to Week 30 as measured by ACR20, ACR50, ACR70, individual ACR parameters (including HAQ-DI), DAS28-CRP, EULAR response, DAS remission and ACR/EULAR remission. No clinically meaningful differences in safety or immunogenicity were found between IXIFI and Remicade.
In line with the findings from the first treatment period, results from the second period with dosing up to Week 54 continued to show the absence of clinically meaningful differences in efficacy, PD, immunogenicity and safety among subjects receiving IXIFI, Remicade, and subjects who transitioned from Remicade to IXIFI.
Overall, in line with the findings from the first 2 treatment periods, results from the third treatment period (final period; from Week 54 to Week 78) supported the efficacy and safety of IXIFI in subjects with moderately to severely active RA who were treated with IXIFI in combination with methotrexate. Furthermore, results from the final period showed the absence of clinically meaningful differences in efficacy, PK, PD, immunogenicity and safety among the three treatment groups in the final period independent of single treatment transition from Remicade to IXIFI at Week 30 or Week 54.
Based on the comparative clinical efficacy and safety results obtained in Study B5371002 in subjects with RA, it is concluded that biosimilarity was demonstrated between IXIFI and Remicade. The totality of evidence supports that IXIFI is biosimilar to Remicade.
Pharmacokinetics: Adults: Single intravenous infusions of 1, 3, 5, 10 or 20 mg/kg of infliximab yielded dose proportional increases in the maximum serum concentration (C
max) and area under the concentration-time curve (AUC). The volume of distribution at steady state (median V
d of 3.0 to 4.1 liters) was not dependent on the administered dose and indicated that infliximab is predominantly distributed within the vascular compartment. No time-dependency of the pharmacokinetics was observed. The elimination pathways for infliximab have not been characterized. Unchanged infliximab was not detected in urine. No major age- or weight-related differences in clearance or volume of distribution were observed in rheumatoid arthritis patients. The pharmacokinetics of infliximab in elderly patients has not been studied. Studies have not been performed in patients with liver or renal disease.
At single doses of 3, 5, or 10 mg/kg, the median C
max values were 77, 118 and 277 micrograms/mL, respectively. The median terminal half-life at these doses ranged from 8 to 9.5 days. In most patients, infliximab could be detected in the serum for at least 8 weeks after the recommended single dose of 5 mg/kg for Crohn's disease and the rheumatoid arthritis maintenance dose of 3 mg/kg every 8 weeks.
Repeated administration of infliximab (5 mg/kg at 0, 2 and 6 weeks in fistulizing Crohn's disease, 3 or 10 mg/kg every 4 or 8 weeks in rheumatoid arthritis) resulted in a slight accumulation of infliximab in serum after the second dose. No further clinically relevant accumulation was observed. In most fistulizing Crohn's disease patients, infliximab was detected in serum for 12 weeks (range 4-28 weeks) after administration of the regimen.
Pediatrics: Infliximab pharmacokinetic characteristics (including peak and trough concentrations and terminal half-life) were generally similar in pediatric (aged 6 to 17 years) and adult patients with Crohn's disease or ulcerative colitis following the administration of 5 mg/kg infliximab.
Population pharmacokinetic analysis based on data obtained from patients with ulcerative colitis (N = 60), Crohn's disease (N = 112), juvenile rheumatoid arthritis (N = 117) and Kawasaki disease (N = 16) with an overall age range from 2 months to 17 years indicated that the total clearance of infliximab did not increase linearly with increasing body weight. As a result, following administration of 5 mg/kg infliximab every 8 weeks, the predicted median steady-state infliximab exposure (area under concentration-time curve at steady state, AUC
ss) in pediatric patients aged 6 to 17 years was approximately 20% lower than the median steady-state drug exposure in adults. The median AUC
ss in pediatric patients aged 2 to less than 6 years was predicted to be approximately 40% lower than that in adults, although the number of patients supporting this estimate is limited.
IXIFI Comparative Pharmacokinetic Studies: The PK similarity of IXIFI and Remicade was evaluated in the clinical development program. Study B5371001 was a three-arm, double-blind, randomized (1:1:1), parallel-group, single-dose study that compared the PK of IXIFI, infliximab-EU and infliximab-US following IV administration of 10 mg/kg to healthy adult subjects.
The 3 study drugs exhibited a similar median PK profile, which was characterized by a rapid increase of serum drug concentrations during each infusion, followed by a multi-phasic decline in drug concentrations after completion of the IV infusion. (See Figure 2.)
Click on icon to see table/diagram/image
The arithmetic mean (±SD) PK parameters for IXIFI, infliximab-EU, and infliximab-US are summarized in Table 8. Consistent with the concentration-time profiles, the mean C
max, AUC
T and AUC
inf estimates were similar among the 3 study drugs. In addition, the inter-subject variability for each of the PK parameters was similar across the 3 study drugs, with %CV values of 20% to 24%, 21% to 25%, and 23% to 28% for C
max, AUC
T and AUC
inf, respectively. (See Table 8.)
Click on icon to see table/diagram/image
The 90% CIs for test-to-reference ratios of C
max, AUC
t, and AUC
inf were all contained within the pre-specified acceptance boundaries of 80.00% to 125.00% for the comparisons of IXIFI to infliximab-US, IXIFI to infliximab-EU, and infliximab-EU to infliximab-US. This study demonstrated the PK similarity of IXIFI to both infliximab-US and infliximab-EU, and of infliximab-EU to infliximab-US. (See Table 9.)
Click on icon to see table/diagram/image
In Study B5371002, the median C
trough and C
max values, as well as the corresponding ranges, were similar between the IXIFI and infliximab-EU arms. The concentrations of serum IXIFI and infliximab-EU were lower in ADA-positive subjects compared to ADA-negative subjects. The effect of ADA on PK in ADA-positive subjects was similar between treatment arms in all three treatment periods. (See Table 10.)
Click on icon to see table/diagram/image
Additionally, a population PK analysis did not reveal any appreciable differences between the PK of infliximab-EU and IXIFI in the RA patient population. This analysis identified covariates of body weight, sex and ADA titers as significant factors influencing infliximab-EU and IXIFI PK. Furthermore, results indicate that the PK of IXIFI was not different between Japanese and non-Japanese patients.
In conclusion, the PK results obtained in Studies B5371001 and B5371002 in healthy subjects and in subjects with RA, respectively, demonstrate PK similarity between IXIFI, infliximab-US, and infliximab-EU.
Toxicology: Preclinical safety data: Conventional preclinical safety data with infliximab is limited. In a developmental toxicity study conducted in mice using an analogous antibody that selectively inhibits the functional activity of mouse TNFα, there was no indication of maternal toxicity, embryotoxicity or teratogenicity. In a fertility and general reproductive function study, the number of pregnant mice was reduced following administration of the same analogous antibody. It is not known whether this finding was due to effects on the males and/or the females. In a 6-month repeated dose toxicity study in mice, using the same analogous antibody against mouse TNFα, crystalline deposits were observed on the lens capsule of some of the treated male mice. No specific ophthalmologic examinations have been performed in patients to investigate the relevance of this finding for humans.
A 6-month study in CD-1 mice was conducted to assess the tumorigenic potential of cV1q anti-mouse TNFα, an analogous antibody. No evidence of tumorigenicity was observed in mice that received intravenous doses of 10 mg/kg or 40 mg/kg cV1q given weekly. The relevance of this study for human risk is unknown.
Long-term studies have not been performed to evaluate the carcinogenic potential of infliximab. Studies in mice deficient in TNFα demonstrated no increase in tumors when challenged with known tumor initiators and/or promoters.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image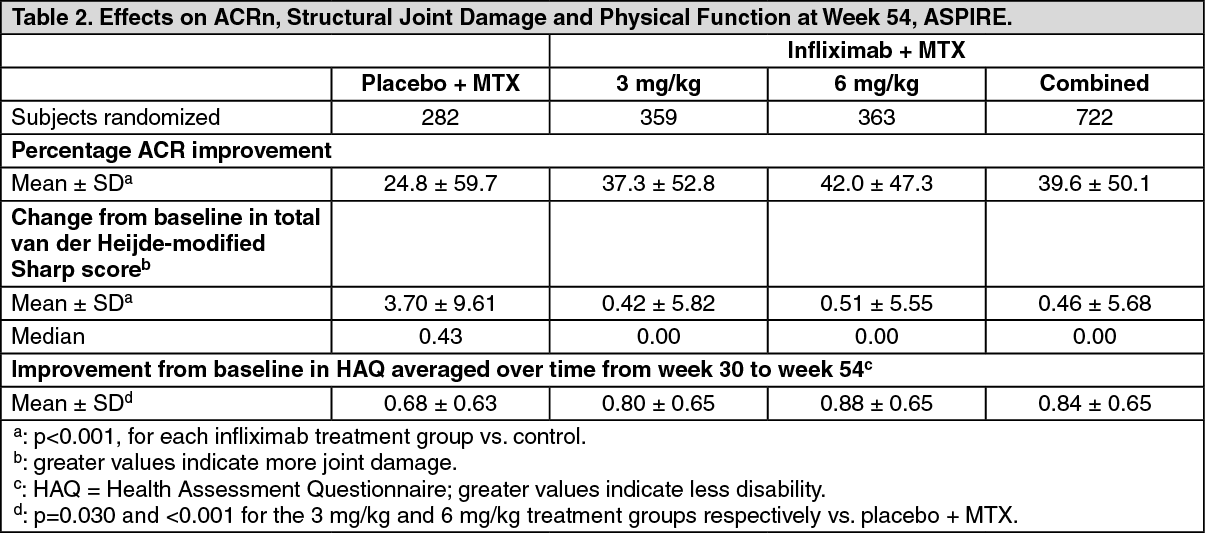 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image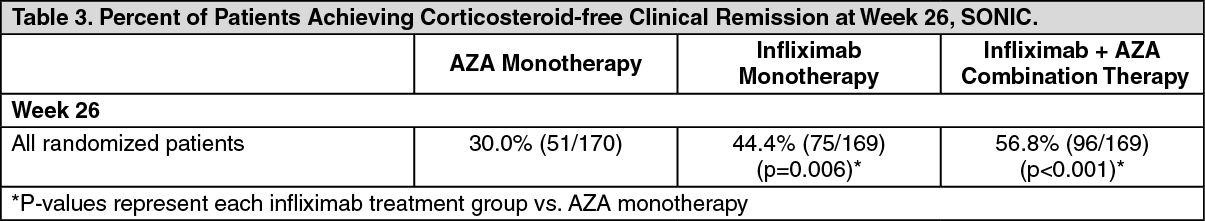 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image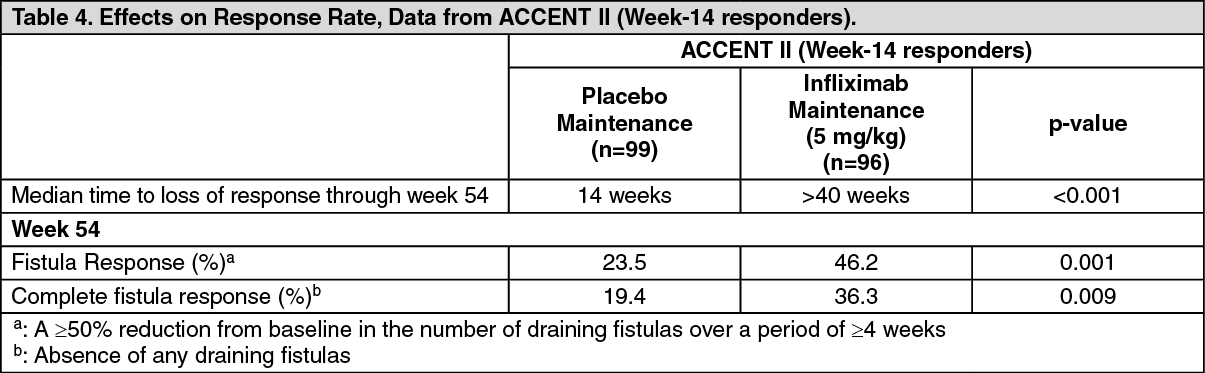 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image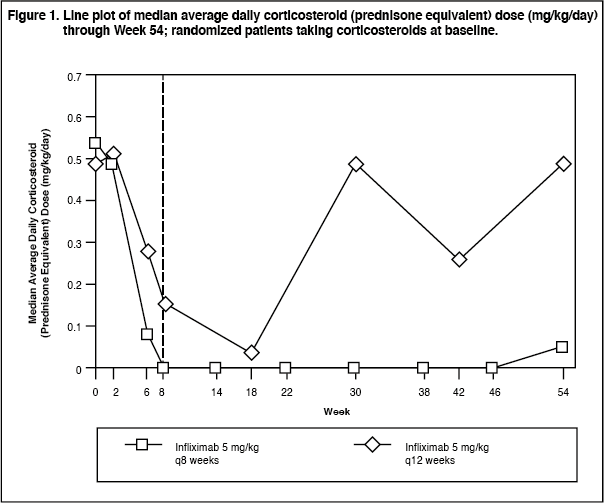 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image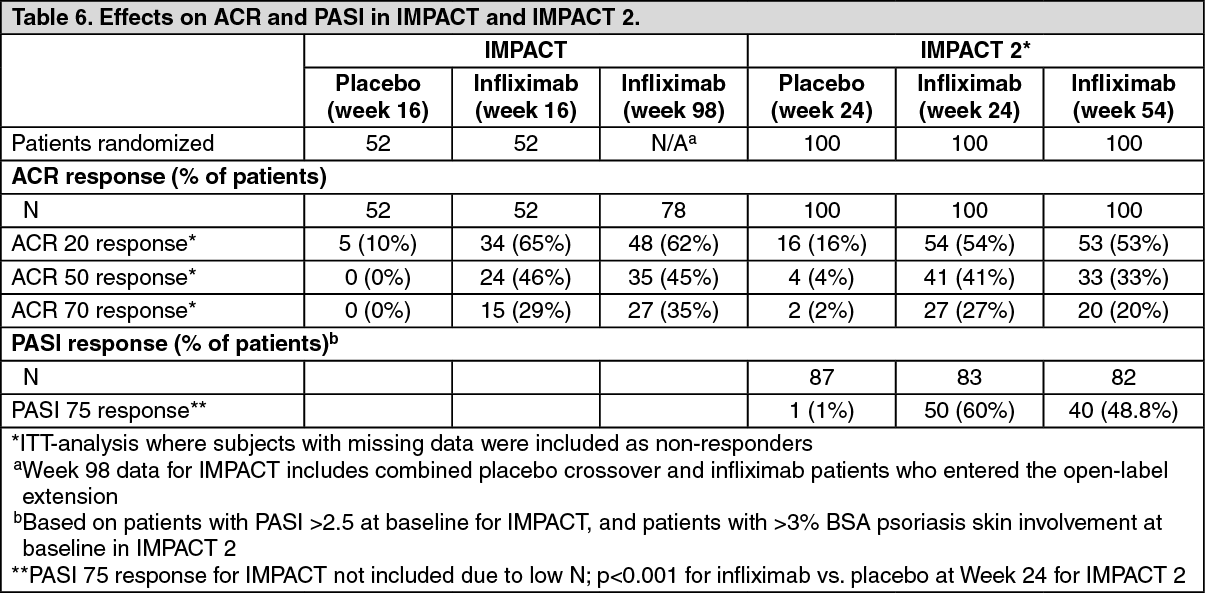 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image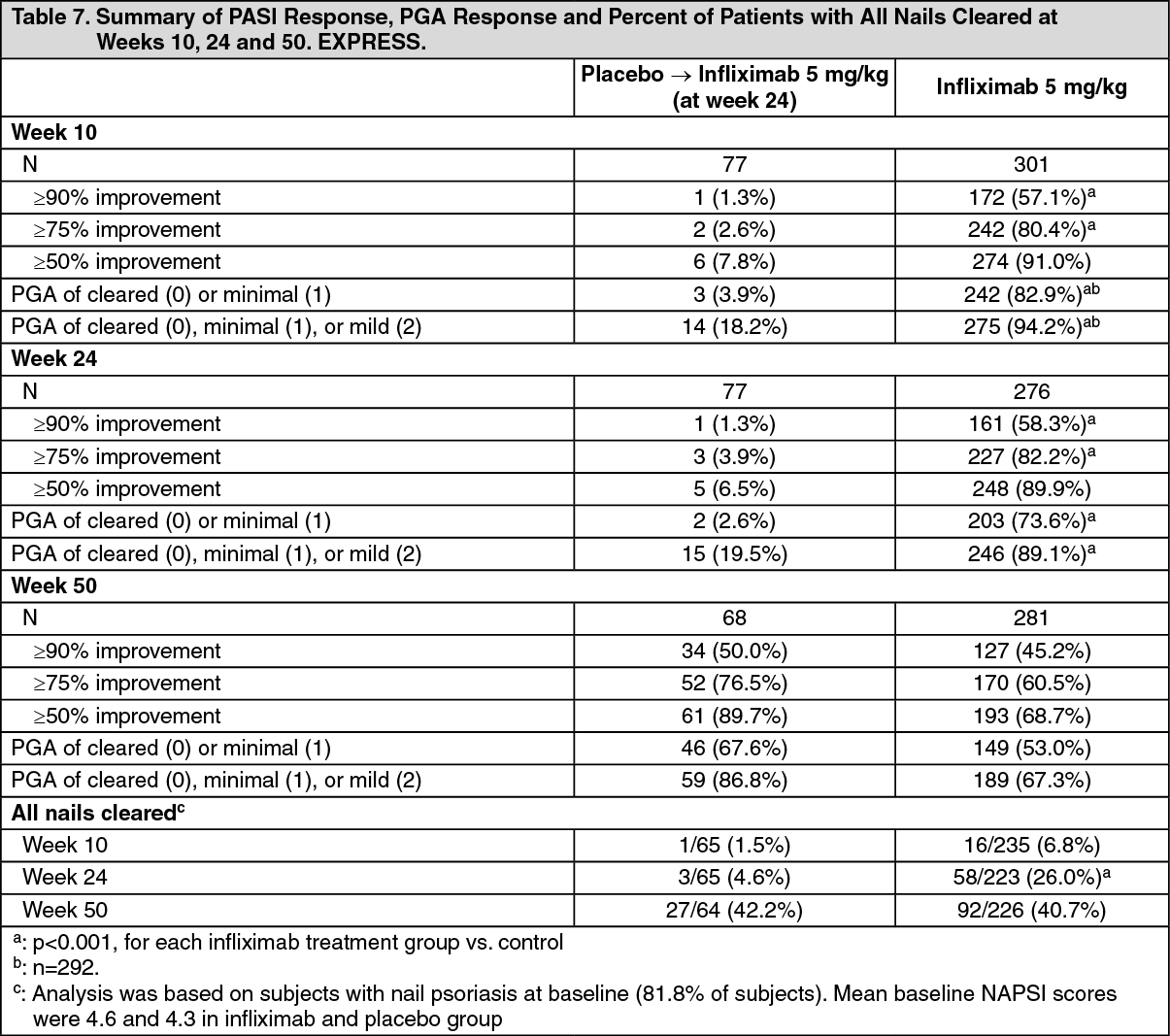 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image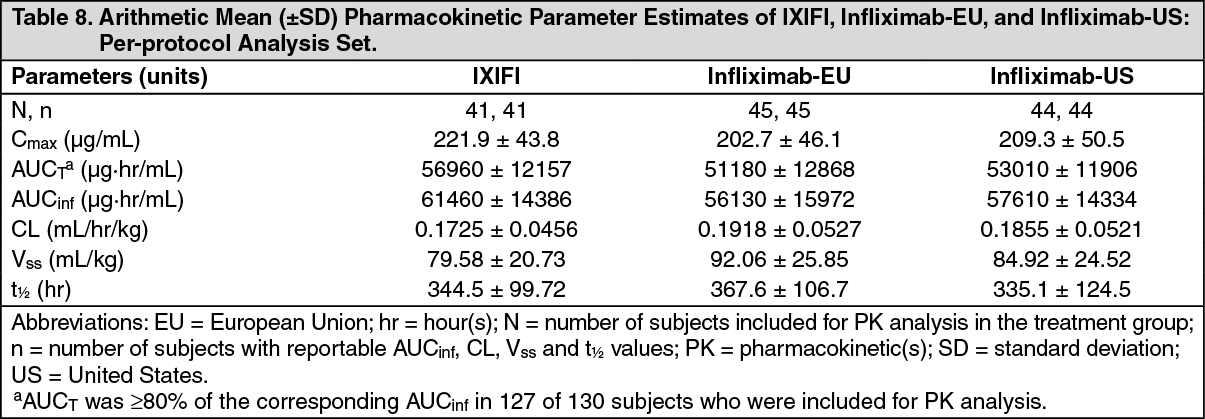 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image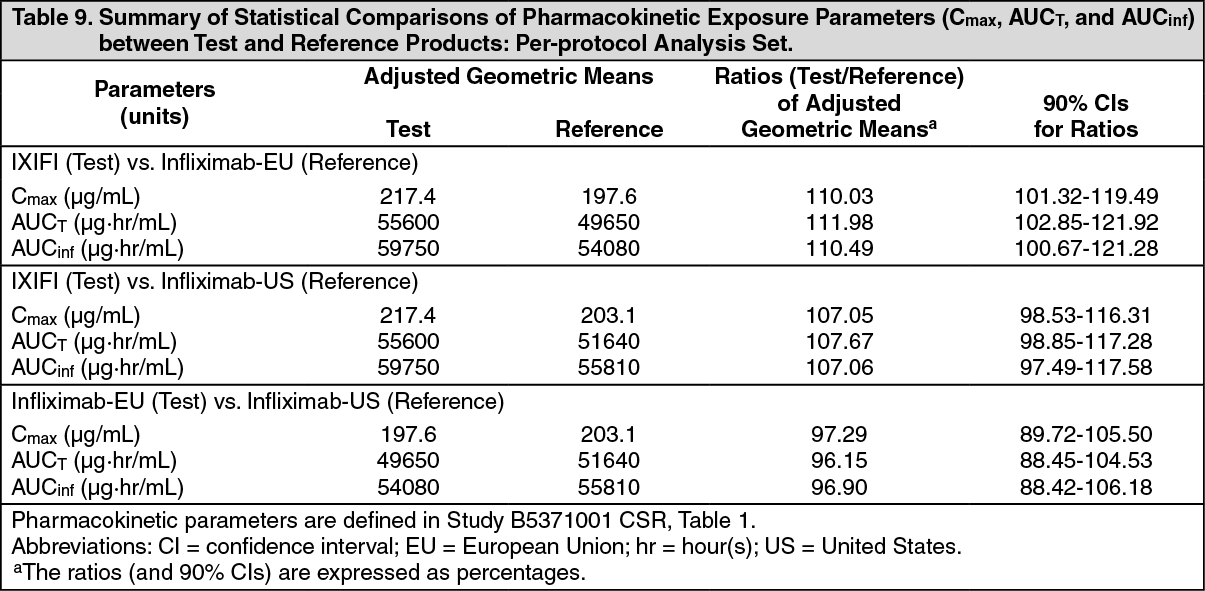 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image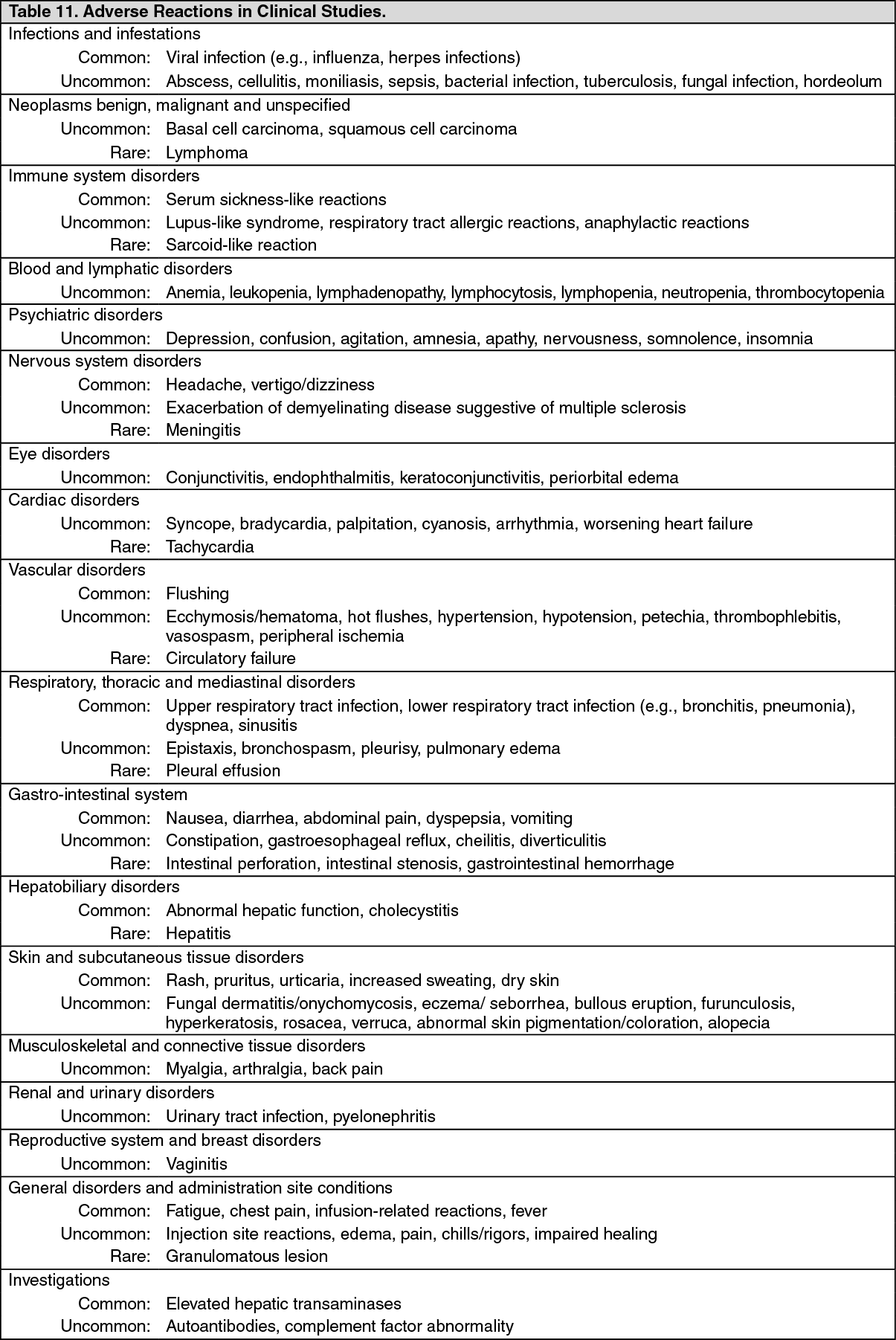 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image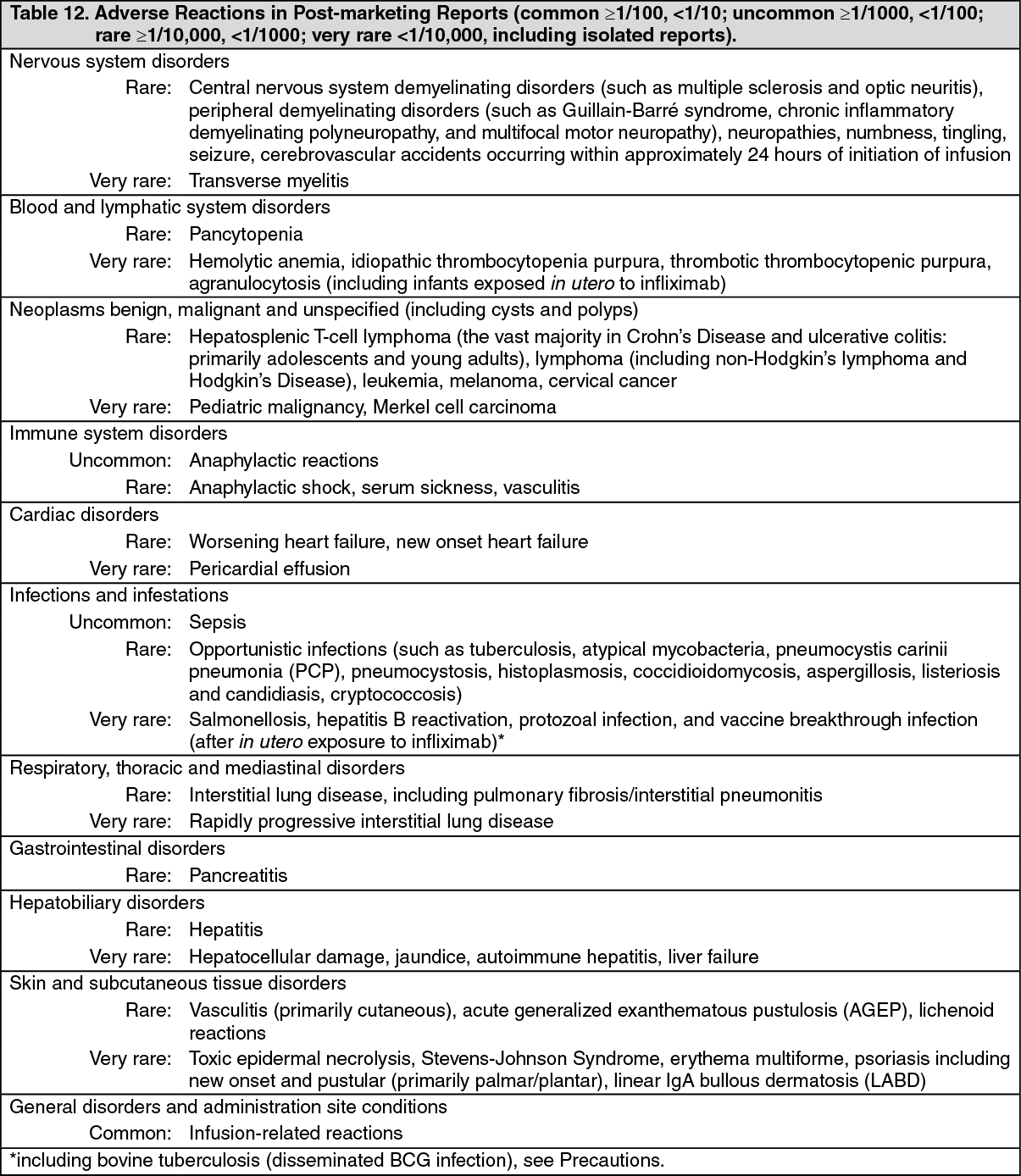 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image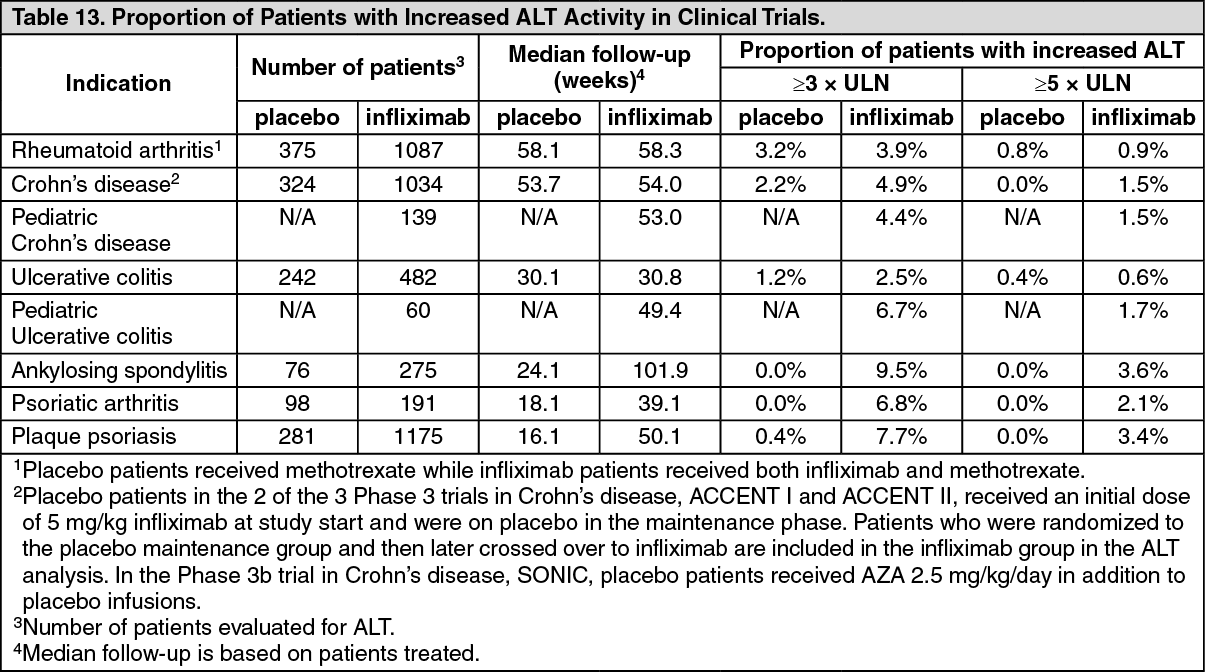 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
