CLINICAL STUDIES: Study Design: The original HYMOVIS R29-09-02 study was a randomized, double-blinded, saline-controlled study conducted at 37 centers in U.S. to evaluate the safety and effectiveness of a two-injection regimen of HYMOVIS in patients with symptomatic osteoarthritis of the knee. This randomized controlled (RC) study was designed to evaluate the safety and effectiveness of a new viscoelastic hydrogel (HYMOVIS) for the treatment of pain associated with symptomatic osteoarthritis of the knee with a 180-day (26 weeks) follow-up with an additional 90-day (12 weeks) open label extension (OLE) phase for the evaluation of safety of one cycle of repeat treatment.
A total of 800 patients were enrolled in the RC study and 529 patients in the OLE study phase. Patients were randomized in a 1:1 ratio to either HYMOVIS or saline injection. The primary endpoint was to determine the superiority of HYMOVIS compared to saline by evaluating the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) in WOMAC VAS Pain Score (WOMAC A, 100mm scale) absolute improvement from baseline at Week 26 (180 days).
Randomized Controlled (RC) Study Phase: For the randomized controlled phase of the original HYMOVIS RC study, a total of 801 were randomized and 800 received treatment at 37 Investigator sites. The time from the first patient enrolled to completion of the last patient visit (last patient out) was approximately 20 months. Individual patient participation lasted approximately nine months (six months for the randomized phase, and three additional months if the patient opted for the OLE phase).
Eligible patients were randomized into one of two treatment groups. The patient and the Evaluator were blinded to the treatment group assignment. The treatment groups were: Group 1: Two intra-articular injections of 3 mL (pre-filled syringes) HYMOVIS (8 mg/mL); one injection on Day 0 and the second on Day 7; Group 2: Two intra-articular injections of 3 mL (pre-filled syringes) of phosphate-buffered saline given on Day 0 and the second on Day 7.
Open Label Extension (OLE) Study Phase: After completion of all safety and efficacy assessments at the Day 180 (Week 26) Visit for the original HYMOVIS RC study, patients were offered the opportunity to further participate in an open label extension phase of the study for the evaluation of safety of one cycle of repeat treatment. The duration of the OLE phase was 90 days beyond receiving repeat treatment.
Eligible patients were summarized based on the injections that they received in the randomized study phase. Treatment groups were as follows: Group 1: Patient received HYMOVIS in the randomized study phase, known as the 2nd HYMOVIS group for the OLE study phase. Two intra-articular injections of 3 mL (pre-filled syringes) HYMOVIS (8 mg/mL) were administered by qualified personnel other than the Blinded Investigator/Observer, at repeat Day 0 and at repeat Day 7.
Group 2: Patients received Placebo in the randomized study phase, known as the 1st HYMOVIS group for the OLE study phase. Two intra-articular injections of 3 mL (pre-filled syringes) HYMOVIS (8 mg/mL) were administered by qualified personnel other than the Blinded Investigator/Observer, at repeat Day 0 and at repeat Day 7.
Results from the OLE study phase were used for the safety profile, but not in the assessment of effectiveness.
Study Population: The patients enrolled in the original RC study were >40 years old and diagnosed with OA of the knee based upon clinical and/or radiographic criteria of the American College of Rheumatology (Kellgren-Lawrence Score II-III) confirmed within three months prior to screening.
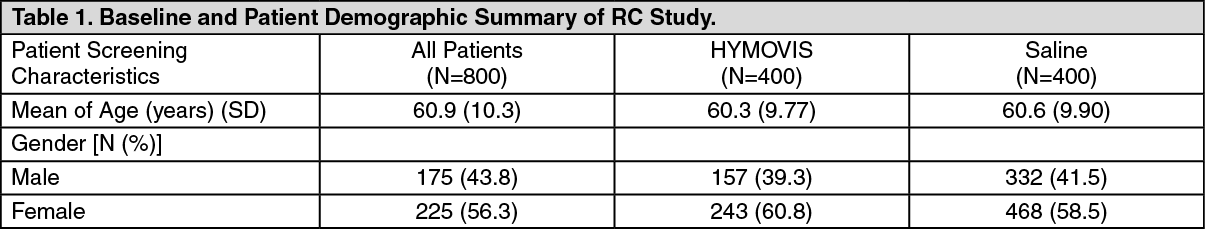
Patient exclusion criteria generally included conditions or medications that could confound the assessment of pain and conditions that could be adversely affected by an intra-articular injection. A total of 800 patients were randomized to either HYMOVIS (n=400) or saline (n=400). These 800 patients comprised the Safety Population (Full Analyses Set). Table 1 summarizes the baseline and patient demographic characteristics for Full Analyses Set population. (See Table 1.)
 Click on icon to see table/diagram/image
Study Treatment and Evaluation Schedule:
Click on icon to see table/diagram/image
Study Treatment and Evaluation Schedule: RC Study Phase: The patients follow up period for the RC Study Phase was 180 Days (26 weeks). Study visits were scheduled for screening, baseline, and Days 7, 14, 28, 60, 90, 120 and 180. Injections were performed at the baseline visit and Day 7 visit. Patients were required to discontinue all analgesics, including NSAIDs prior to the baseline visit and to accept "rescue" acetaminophen as the only medication for treatment of joint pain during the study. "Rescue" medication was not permitted within 24 hours of any study visit.
OLE Study Phase: During the Open-Label Extension Study Phase the follow up visits were scheduled at Day 7, Day 14 and Day 90 following first injection of the retreatment cycle.
SAFETY RESULTS: Safety analyses were performed for this study on the safety population, which was defined as all randomized patients of the original RC Study Phase and the Open Label Extension (OLE) Study Phase. Treatment-emergent AEs were summarized by treatment group and categorized by severity and relationship to the study procedures.
To assess the safety of a repeat injection regimen of two 3 mL of HYMOVIS the compliant patients from both arms were permitted to enter a 90 day open-label repeat treatment phase after the completion of the initial study injection regimen.
RC Study Phase: A summary of Adverse Events recorded in the RC Study Phase is presented in Table 2 as follows. (See Table 2 and Table 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The treatment-emergent adverse events (AEs) most frequently reported are recorded in Table 4 as follows. Adverse Events were considered typical of viscosupplementation injections in this patient population and mostly were mild or moderate in severity. (See Table 4.)
Click on icon to see table/diagram/image
Open-Label Extension (OLE) Study Phase: A summary of Adverse Events recorded in the OLE Study Phase is presented in Table 5 as follows. (See Table 5.)
Click on icon to see table/diagram/image
EFFECTIVENESS RESULTS: RC Study Phase: PRIMARY EFFECTIVENESS ENDPOINT: The analysis of the effectiveness of HYMOVIS was based on the modified Full Analysis Set (mFAS) population (n=786 patients) evaluable at the 6-month time point. The pain reduction from baseline for HYMOVIS was -19.47 mm on the whole 100 mm WOMAC A Pain scale and that of saline placebo was -18.13 mm. The primary effectiveness endpoint was not met in this study. As shown as follows in Table 6, the study did not demonstrate a statistically significant difference, as well as a clinically meaningful difference of at least 6 mm, between the two groups in WOMAC A Pain Scores at six months. (See Table 6.)
Click on icon to see table/diagram/image
The analysis was based on a two sided t test at 180 days for the primary endpoint.
SECONDARY EFFECTIVENESS ENDPOINTS: All secondary endpoints shown as follows were not statistically different from Saline placebo: Responder, based on OMERACT-OARSI*, at 26 weeks; Function measured in WOMAC C; VAS pain measured in WOMAC A 1 (Pain subscore); WOMAC Global Score; Stiffness measured in WOMAC B.
*Outcome Measures in Rheumatology Clinical Trials and Osteoarthritis Research Society International (OMERACT-OARSI) criteria of response
HYMOVIS vs. HYALGAN (Sodium Hyaluronate) Post Hoc Non-Inferiority Analysis: The primary effectiveness endpoint for the HYMOVIS pivotal RC study (R29-09-02), comparison of the reductions in the WOMAC Pain Score (WOMAC A) from baseline through 180 days, was used for a post-hoc non-inferiority comparison of HYMOVIS to HYALGAN, previously approved under P950027 for an identical indication for use. WOMAC A Pain Scores were utilized to determine the non-inferiority of HYMOVIS to HYALGAN using Bayesian regression analysis. Under this Bayesian analysis, a two-injection treatment regimen of HYMOVIS was assessed for its ability to provide pain relief non-inferior to that of a 5-injection treatment regimen of HYALGAN as determined through comparison of the reduction in WOMAC A Pain Scores from baseline through 180 days utilizing a non-inferiority margin of 5 mm on the 100mm WOMAC A Pain Scale.
The primary effectiveness endpoint for this non-inferiority analysis was met as calculated using a Bayesian regression analysis with posterior probability of 97%.
Non-Inferiority Endpoints: The non-inferiority margins were set conservatively at Δ=5 mm (on a 100mm WOMAC VAS Scale), 10 mm for Patient Global Assessment, and a 0.8 relative risk for the OMERACT-OARSI response rate.
The mean differences between treatment groups are calculated and a lower one-sided 97.5% confidence interval is constructed. If the lower bound is greater than -Δ, then 'Non-inferiority' is obtained for HYMOVIS relative to the five-injection HYALGAN group.
Clinical Significance Demonstration: To demonstrate clinical significance a cumulative distribution method for determining of the change from baseline for each of the endpoints was employed. Cumulative Distribution Function (CDF) plots comparing the HYMOVIS two injection regimen to the HYALGAN five-injection regimen effectiveness were conducted and provided for primary and secondary endpoints. At -6.0 mm on a 100mm WOMAC VAS scale, which is considered by Agency a valid clinically important difference, the CDF plots demonstrate that HYMOVIS demonstrates a higher degree of clinical improvement than HYALGAN for all significant test endpoints.
Figures 1 and 2 as follows show the Cumulative Distribution Plot for Change in WOMAC A Pain Score from Baseline to Day 120 and Day 180. (See Figure 1.)
Click on icon to see table/diagram/image
The CDF curves for the endpoints (WOMAC Pain Score at day 180) show that the HYMOVIS mPP population demonstrates a higher degree of clinical improvement at day 180 to HYALGAN. (See Figure 2.)
Click on icon to see table/diagram/image
The CDF curves for the endpoints (WOMAC Pain Score at day 120) show that the HYMOVIS population demonstrates a higher degree of clinical improvement at day 120.
BENEFIT-RISK ANALYSIS: Two injections of HYMOVIS provide a benefit in pain reduction in patients with osteoarthritis in the knee that is non-inferior to the pain reduction provided by 5 injections of HYALGAN, a product previously approved for the same indications. Safety assessment results support a favorable benefit/risk ratio; that is, the probable benefits outweigh the probable risks of transitory adverse events such as pain in the treatment of osteoarthritis of the knee in patients who have failed to adequately respond to conservative non-pharmacological therapy and simple analgesics (e.g., acetaminophen).



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image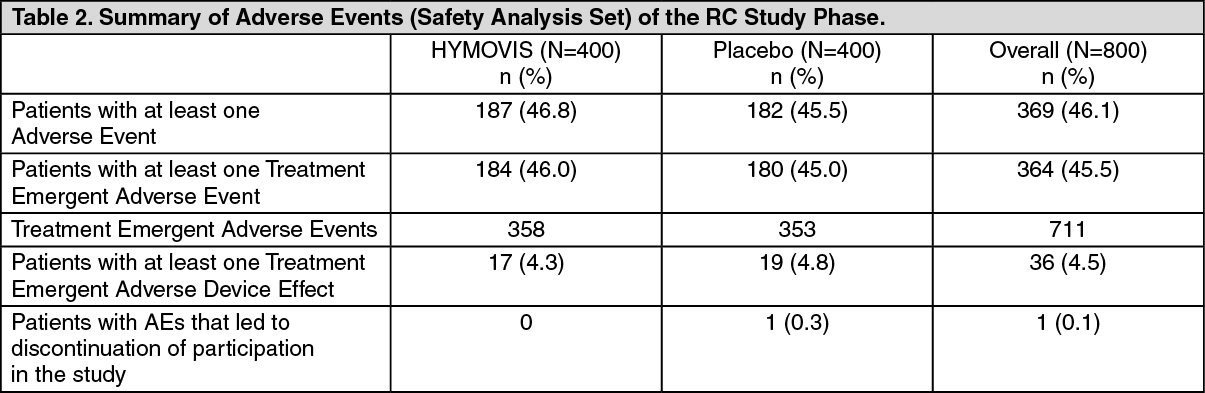 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image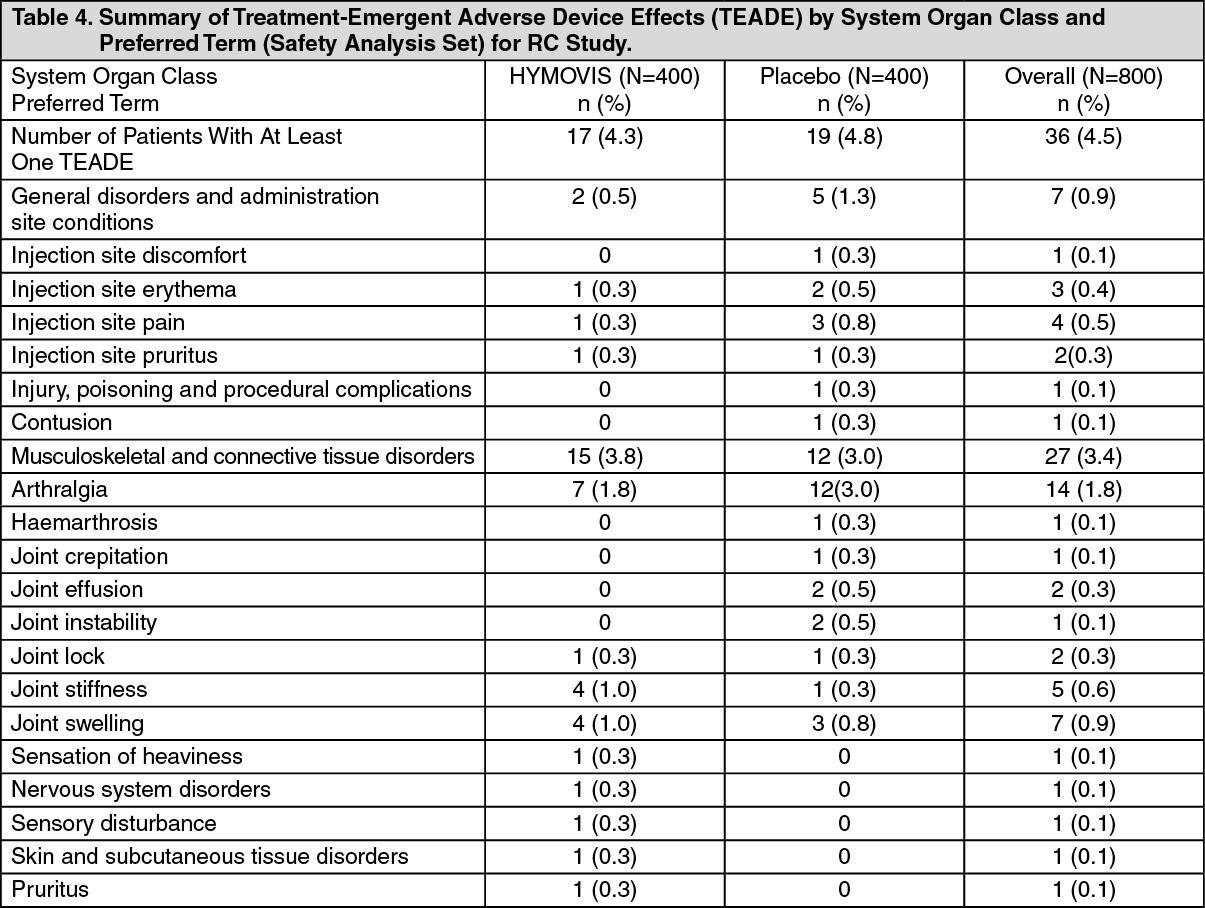 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image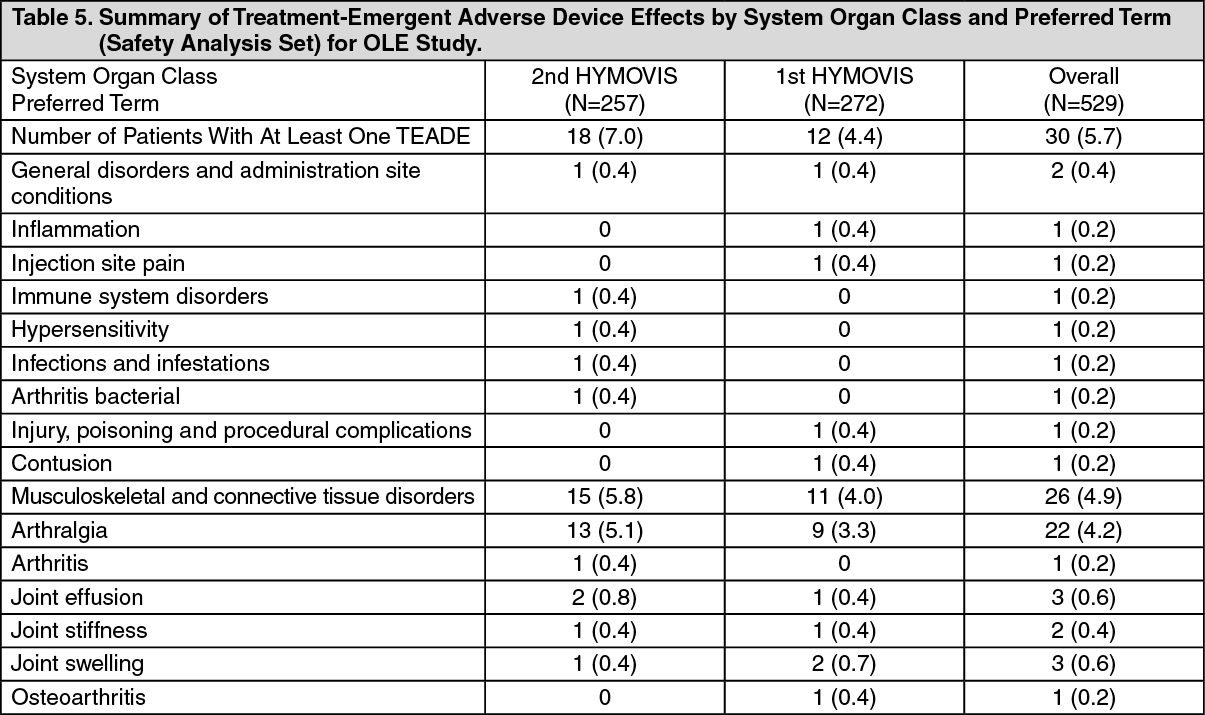 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image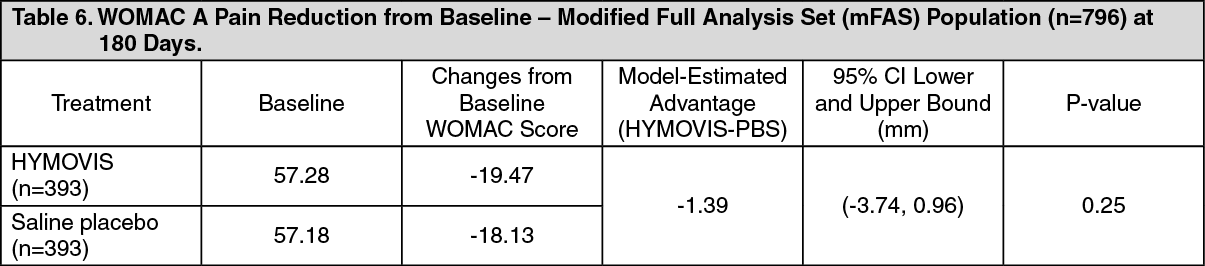 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image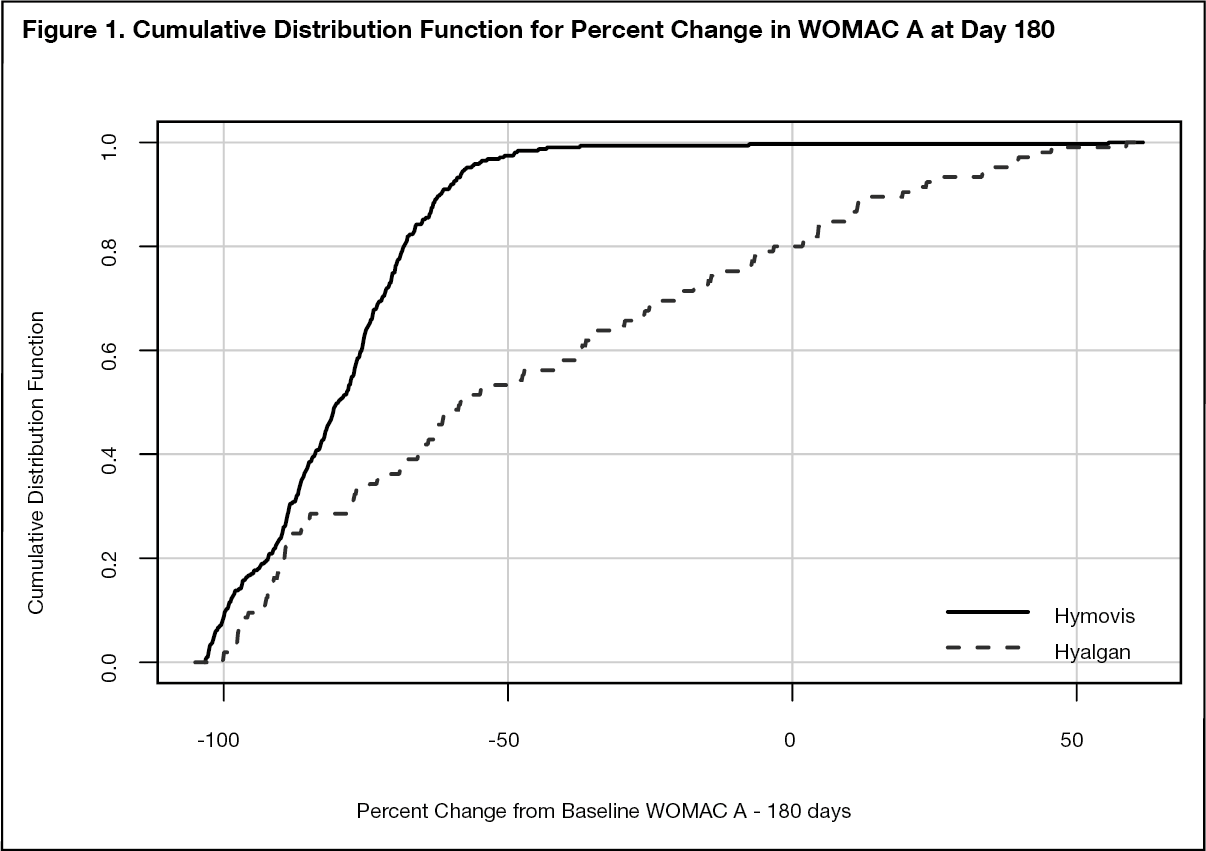 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image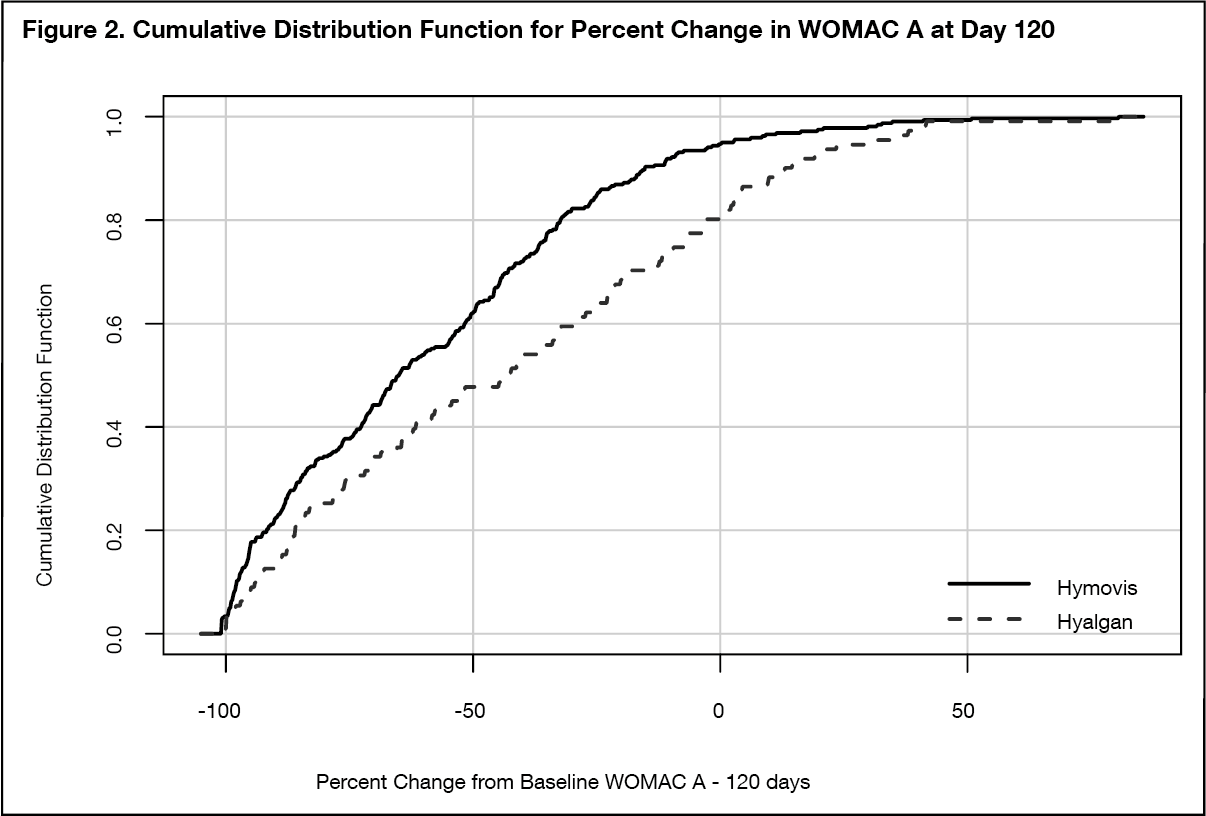 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
