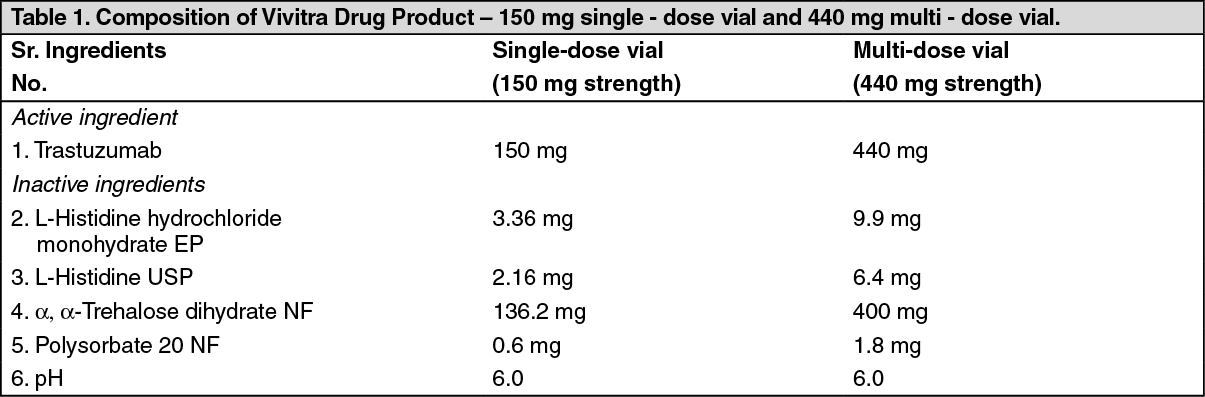
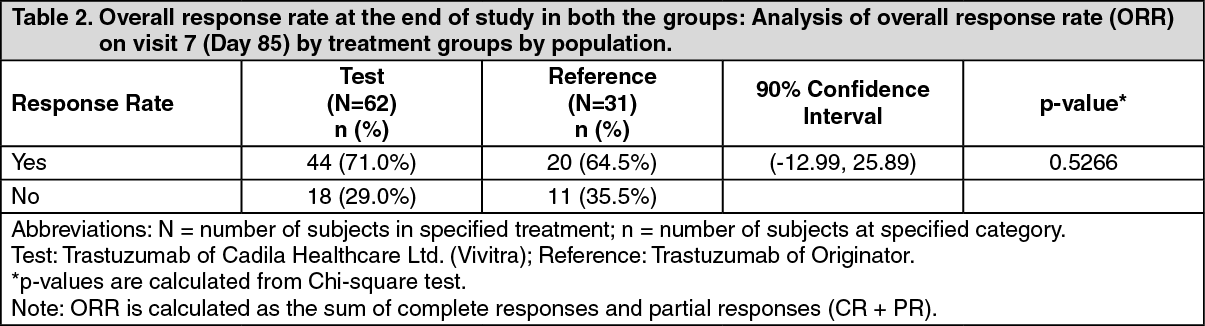
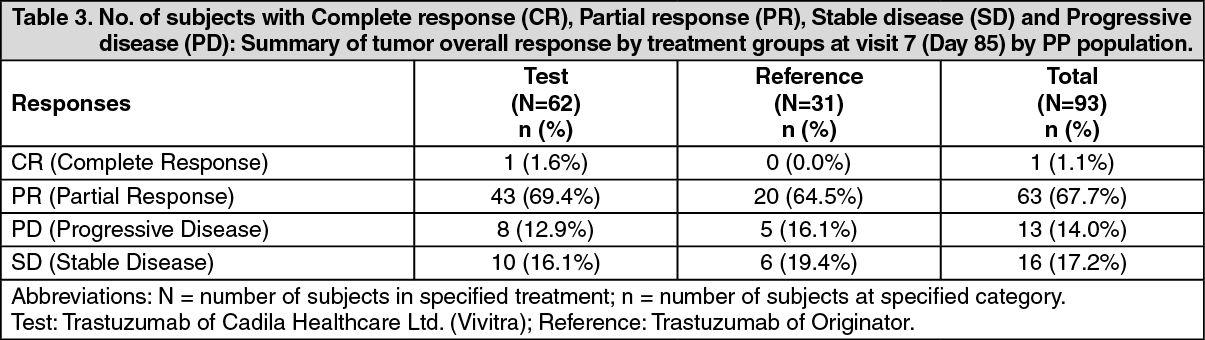
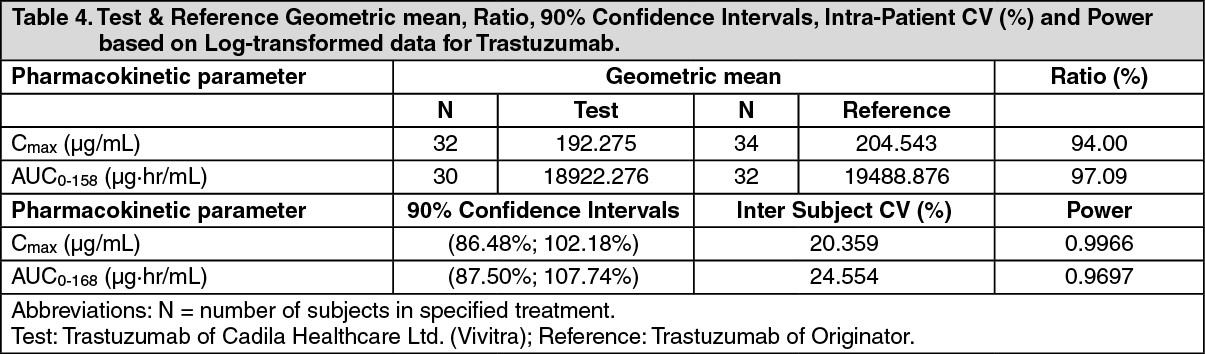
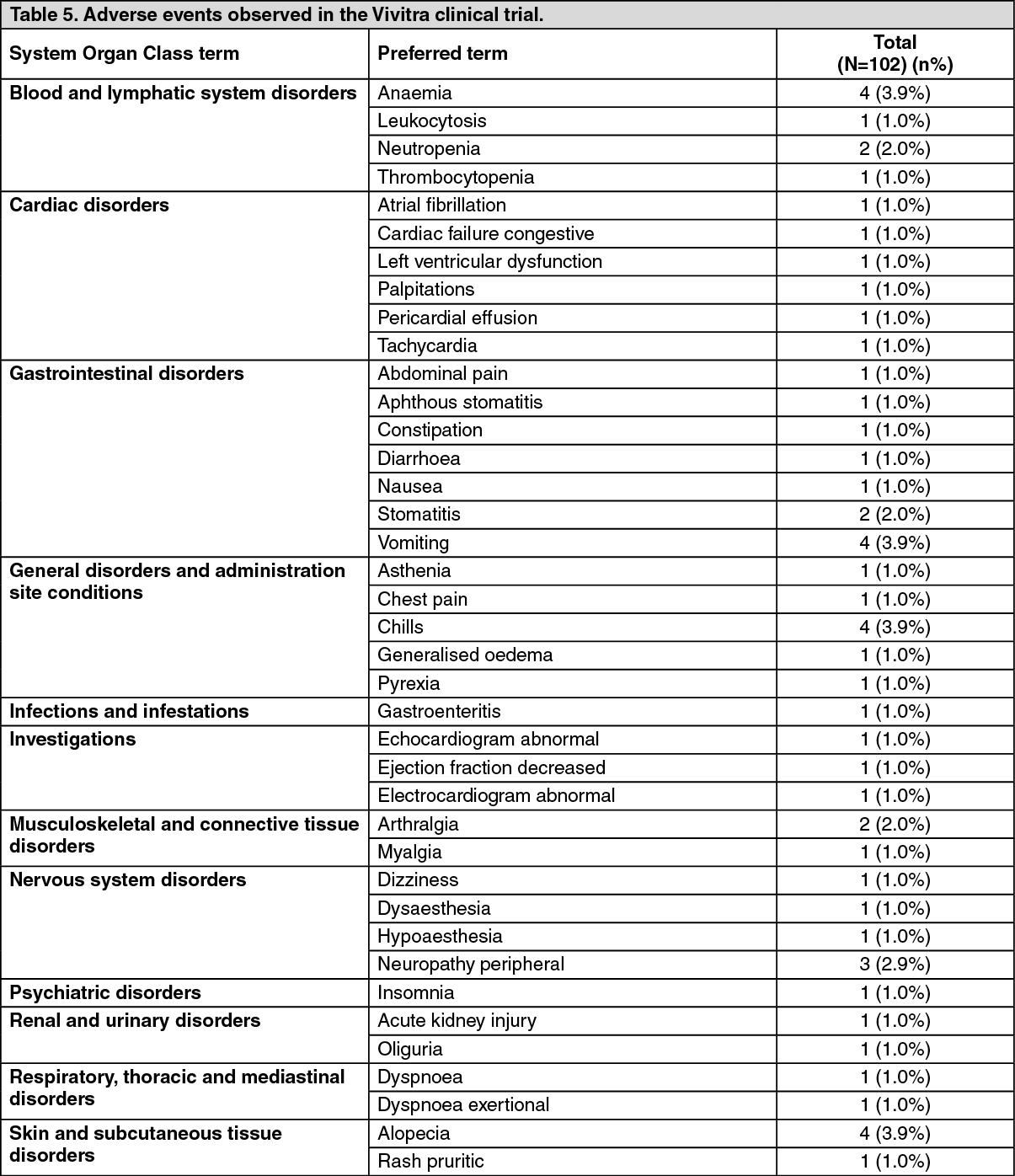
ADVERSE EVENTS OBSERVED IN THE Vivitra CLINICAL TRIAL WERE: See Table 5.
 Click on icon to see table/diagram/image
ADVERSE EVENTS REPORTED WITH THE USE OF TRASTUZUMAB:
Click on icon to see table/diagram/image
ADVERSE EVENTS REPORTED WITH THE USE OF TRASTUZUMAB: Following adverse events reported with the use of intravenous Trastuzumab alone or in combination with chemotherapy:
General disorders and administration site conditions: Asthenia, Influenza-like symptoms, Infusion related reaction, Chest pain, Chills, Pain, Fatigue, Mucosal inflammation, Pyrexia, Peripheral oedema, Oedema, Malaise.
Blood and lymphatic system disorders: Anaemia, Neutropenia, Febrile neutropenia, Thrombocytopenia, White blood cell count decreased/leukopenia, Hypoprothrombinaemia.
Infections and infestations: Infection, Neutropenic sepsis, Nasopharyngitis, Herpes zoster, Sinusitis, Influenza, Cystitis, Cellulitis, Upper respiratory tract infection, Urinary tract infection, Rhinitis, Sepsis, Pharyngitis, Erysipelas, Skin infection.
Cardiac disorders: Palpitation, Cardiac flutter, Heart beat irregular, Ejection fraction decreased, Blood pressure decreased, Blood pressure increased, Cardiac failure (congestive), Supraventricular tachyarrhythmia, Pericardial effusion, Cardiomyopathy, Cardiogenic shock, Pericarditis, Bradycardia, Gallop rhythm present.
Vascular disorders: Hot flush, Vasodilatation, Hypotension.
Respiratory, thoracic and mediastinal disorders: Cough, Wheezing, Epistaxis, Dyspnoea, Rhinorrhoea, Pneumonia, Pleural effusion, Asthma, Lung disorder, Pneumonitis, Pulmonary fibrosis, Respiratory failure, Respiratory distress, Lung infiltration, Acute pulmonary oedema, Bronchospasm, Acute respiratory distress syndrome, Hypoxia, Interstitial lung disease, Orthopnoea, Pulmonary oedema, Laryngeal oedema, Oxygen saturation decreased.
Gastrointestinal disorders: Nausea, Diarrhoea, Vomiting, Lip swelling, Abdominal pain, Dyspepsia, Stomatitis, Constipation, Pancreatitis, Dry mouth, Haemorrhoids.
Hepatobiliary disorders: Liver tenderness, Hepatocellular injury, Hepatitis, Jaundice, Hepatic failure.
Metabolism and nutrition disorders: Anorexia, Weight decreased/Weight loss, Hyperkalaemia.
Immune system disorders: Hypersensitivity, Anaphylactic shock, Anaphylactic reaction.
Psychiatric disorders: Insomnia, Depression, Thinking abnormal, Anxiety.
Nervous system disorders: Tremor, Paraesthesia, Dysgeusia, Dizziness, Headache, Ataxia, Peripheral neuropathy, Hypertonia, Somnolence, Paresis, Brain oedema.
Ear and labyrinth disorders: Deafness.
Eye disorders: Lacrimation increased, Conjunctivitis, Dry eye, Retinal haemorrhage, Papilloedema.
Musculoskeletal and connective tissue disorders: Muscle tightness, Myalgia, Arthralgia, Arthritis, Bone pain, Back pain, Pain in extremity, Neck Pain, Muscle spasms.
Skin and subcutaneous tissue disorders: Erythema, Alopecia, Rash, Swelling face, Palmar-plantar erythrodysaesthesia syndrome, Nail disorder, Acne, Ecchymosis, Dry skin, Dermatitis, Onychoclasis, Maculopapular rash, Hyperhidrosis, Pruritus, Urticaria, Angioedema.
Renal and urinary disorders: Renal disorder, Renal failure, Glomerulonephritis membranous, Glomerulonephropathy.
Reproductive system and breast disorders: Breast inflammation/mastitis.
Pregnancy, puerperium and perinatal conditions: Pulmonary hypoplasia, Oligohydramnios, Renal hypoplasia.
Injury, poisoning and procedural complications: Contusion.
Neoplasms benign, malignant and unspecified (incl. Cysts and polyps): Neoplasm progression, malignant neoplasm progression.



 Sign Out
Sign Out