Pharmacology: Pharmacodynamics: Rabeprazole: Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H
2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H
+, K
+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. In gastric parietal cells, rabeprazole is protonated, accumulates and is transformed to an active sulfonamide. When studied
in vitro, rabeprazole is chemically activated at pH 1.2 with a half-life (t
½) of 78 sec. It inhibits acid transport in porcine gastric vesicles with a t
½ of 90 sec.
Antisecretory Activity: The antisecretory effect begins within 1 hr after oral administration of rabeprazole 20 mg. The median inhibitory effect of rabeprazole on 24 hr gastric acidity is 88% of maximal after the first dose. Rabeprazole 20 mg inhibits basal and peptone meal-stimulated acid secretion vs placebo by 86% and 95%, respectively, and increases the percent of a 24-hr period that the gastric pH>3 from 10-65% (see Table 1). This relatively prolonged pharmacodynamic action compared to the short pharmacokinetic t
½ (1-2 hr) reflects the sustained inactivation of the H
+, K
+ATPase.
Gastric Acid Parameters: Rabeprazole versus placebo after 7 days of once-daily dosing: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Compared to placebo, rabeprazole 10 mg, 20 mg and 40 mg administered once daily for 7 days significantly decreased intragastric acidity with all doses for each of 4 meal-related intervals and the 24-hr time period overall. In this study, there were no statistically significant differences between doses; however; there was a significant dose-related decrease in intragastric acidity.
Effects on Esophageal Acid Exposure: In patients with gastroesophageal reflux disease (GERD) and moderate to severe esophageal acid exposure, rabeprazole 20- and 40 mg/day decreased 24-hr esophageal acid exposure. After 7 days of treatment, the percentage of time that esophageal pH <4 decreased from baselines of 24.7% for 20 mg and 23.7% for 40 mg, to 5.1% and 2%, respectively. Normalization of 24-hr intraesophageal acid exposure was correlated to gastric pH >4 for at least 35% of the 24-hr period; this level was achieved in 90% of subjects receiving rabeprazole 20 mg and in 100% of subjects receiving rabeprazole 40 mg. With rabeprazole 20- and 40 mg/day, significant effects on gastric and esophageal pH were noted after 1 day of treatment and more pronounced after 7 days of treatment.
Effects on Serum Gastrin: In patients given daily doses of rabeprazole for up to 8 weeks to treat ulcerative or erosive esophagitis and in patients treated for up to 52 weeks to prevent recurrence of disease, the median fasting gastrin level increased in a dose-related manner. The group median values stayed within the normal range.
In a group of subjects treated daily with rabeprazole 20 mg for 4 weeks, a doubling of mean serum gastrin concentrations were observed. Approximately 35% of these treated subjects developed serum gastrin concentrations above the upper limit of normal.
Effects on Enterochromaffin-Like (ECL) Cells: Increased serum gastrin secondary to antisecretory agents stimulates proliferation of gastric ECL cells which, over time, may result in ECL cell hyperplasia in rats and mice and gastric carcinoids in rats, especially in females.
In over 400 patients treated with rabeprazole, (10 or 20 mg/day) for up to 1 year, the incidence of ECL cell hyperplasia increased with time and dose, which is consistent with the pharmacological action of the proton-pump inhibitor. No patient developed the adenomatoid, dysplastic or neoplastic changes of ECL cells in the gastric mucosa. No patient developed the carcinoid tumors observed in rats.
Endocrine Effects: Studies in humans for up to 1 year have not revealed clinically significant effects on the endocrine system. In healthy male volunteers treated with rabeprazole for 13 days, no clinically relevant changes have been detected in the following endocrine parameters examined: 17β-estradiol, thyroid-stimulating hormone, triiodothyronine, thyroxine, thyroxine-binding protein, parathyroid hormone, insulin, glucagon, renin, aldosterone, follicle-stimulating hormone, luteotrophic hormone, prolactin, somatotrophic hormone, dehydroepiandrosterone, cortisol-binding globulin and urinary 6β-hydroxycortisol, serum testosterone and circadian cortisol profile.
Other Effects: In humans treated with rabeprazole for up to 1 year, no systemic effects have been observed on the central nervous, lymphoid, hematopoietic, renal, hepatic, cardiovascular or respiratory systems. No data are available on long-term treatment with rabeprazole and ocular effects.
Itopride: Itopride promotes gastrointestinal motility through synergism of its dopamine D2-receptor antagonistic action and its acetylcholinesterase-inhibitory action. In addition to these actions, itopride has an antiemetic action, which is based on its dopamine D2-receptor antagonistic action at chemoreceptor trigger zone (CTZ).
Pharmacokinetics: Rabeprazole: Rablet-I tablets are enteric-coated to allow rabeprazole sodium, which is acid labile to pass through the stomach relatively intact. After oral administration of rabeprazole 20 mg, peak plasma concentrations (C
max)
of rabeprazole occur over a range of 2-5 hrs (T
max). The rabeprazole C
max and AUC are linear over an oral dose range of 10-40 mg. There is no appreciable accumulation when doses of 10-40 mg are administered every 24 hrs; the pharmacokinetics of rabeprazole is not altered by multiple dosing.
The plasma half-life (t
½) ranges from 1-2 hrs.
Absorption: Absolute bioavailability for rabeprazole 20 mg oral tablet (compared to IV administration) is approximately 52%. Rabeprazole may be taken without regard to timing of meals.
Distribution: Rabeprazole is 96.3% bound to human plasma proteins.
Metabolism: Rabeprazole is extensively metabolized. The thioether and sulphone are the primary metabolites measured in human plasma.
In vitro studies have demonstrated that rabeprazole is metabolized in the liver primarily by cytochromes P450 3A (CYP3A) to a sulphone metabolite and cytochrome P450 2C19 (CYP2C19) to desmethyl rabeprazole. The thioether metabolite is formed non-enzymatically by reduction of rabeprazole.
Excretion: Following a single-oral dose of
14C-labeled rabeprazole 20 mg, approximately 90% of the drug was eliminated in the urine, primarily as thioether carboxylic acid; its glucuronide and mercapturic acid metabolites. The remainder of the dose was recovered in the feces. No unchanged rabeprazole was recovered in the urine or feces.
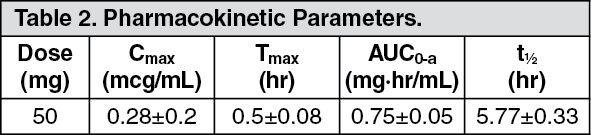
Itopride: Serum Concentrations: The serum concentrations and pharmacokinetic parameters in healthy adults, after single oral administration of itopride hydrochloride 50 mg in the fasting state, are shown in Table 2.
Click on icon to see table/diagram/image
Distribution: Results of Animal Experiments: The concentrations reached the maximum in almost all tissues at 1-2 hrs after a single oral dose of 5 mg/kg of C-itopride hydrochloride to rats and the level at 2 hrs after administration was high in the kidneys, small intestines, liver, adrenal glands and stomach in order of the level (high-low), and the transfer into the central nervous system eg, brain and spinal marrow, was minimal.
In intraduodenal administration of 5 mg/kg of C-itopride hydrochloride to rats, the radioactivity concentrations in the gastric muscular layers were about 2 times as high as those in the blood.
Metabolism and Excretion: At a single dose of itopride hydrochloride 100 mg was orally administered to healthy adults (6 men) in the fasting state. Urinary excretion rate within 24 hrs after administration was highest in the N-oxide form [67.54% of the dose (89.41% of the urinary excretion)] and in the unchanged compound was the second (4.14%) and in the others were minimal.
In the experiments using microsomes that express a human CYP or flavine monooxygenase (FMO), it was found that FMO1 and FMO3 were involved in the production of main metabolite in oxide form. However, no N-oxygenase activity of CYP1A2, -2A6, -2B6, -2C8, -2C9, 2C19, 2D6, 2E1 or 3A4 was detected.



 Sign Out
Sign Out