Pharmacology: Pharmacodynamics: Mode of action: Voriconazole is a triazole antifungal agent. The primary mode of action of voriconazole is the inhibition of fungal cytochrome P-450 mediated 14 alpha-lanosterol demethylation, an essential step in fungal ergosterol biosynthesis. The accumulation of 14 alpha-methyl sterols correlates with the subsequent loss of ergosterol in the fungal cell membrane and may be responsible for the antifungal activity of voriconazole. Voriconazole has been shown to be more selective for fungal cytochrome P-450 enzymes than for various mammalian cytochrome P-450 enzyme systems.
Pharmacokinetic/Pharmacodynamic relationship: In 10 therapeutic studies, the median for the average and maximum plasma concentrations in individual subjects across the studies was 2,425 ng/mL (inter-quartile range 1,193 to 4,380 ng/mL) and 3,742 ng/mL (inter-quartile range 2,027 to 6,302 ng/mL), respectively. A positive association between mean, maximum or minimum plasma voriconazole concentration and efficacy in therapeutic studies was not found.
Pharmacokinetic-pharmacodynamic analyses of clinical trial data identified positive associations between plasma voriconazole concentrations and both liver function test abnormalities and visual disturbances.
Clinical Experience: Successful outcome in this section is defined as complete or partial response.
Aspergillus infections - efficacy in aspergillosis patients with poor prognosis: Voriconazole has
in vitro fungicidal activity against
Aspergillus spp. The efficacy and survival benefit of voriconazole compared to conventional amphotericin B in the primary treatment of acute invasive aspergillosis was demonstrated in an open, randomised, multicenter study in 277 immunocompromised patients treated for 12 weeks. Voriconazole was administered intravenously with a loading dose of 6 mg/kg every 12 hours for the first 24 hours followed by a maintenance dose of 4 mg/kg every 12 hours for a minimum of seven days. Therapy could then be switched to the oral formulation at a dose of 200 mg every 12 hours. Median duration of IV voriconazole therapy was 10 days (range 2-85 days). After IV voriconazole therapy, the median duration of PO voriconazole therapy was 76 days (range 2-232 days).
A satisfactory global response (complete or partial resolution of all attributable symptoms, signs, radiographic/bronchoscopic abnormalities present at baseline) was seen in 53% of voriconazole-treated patients compared to 31% of patients treated with comparator. The 84-day survival rate for voriconazole was statistically significantly higher than that for the comparator and a clinically and statistically significant benefit was shown in favor of voriconazole for both time to death and time to discontinuation due to toxicity.
This study confirmed findings from an earlier, prospectively designed study where there was a positive outcome in subjects with risk factors for a poor prognosis, including graft versus host disease, and, in particular, cerebral infections (normally associated with almost 100% mortality).
The studies included cerebral, sinus, pulmonary and disseminated aspergillosis in patients with bone marrow and solid organ transplants, hematological malignancies, cancer and AIDS.
Serious invasive Candida infections - efficacy in non-neutropenic patients:
The efficacy of voriconazole compared to the regimen of amphotericin B followed by fluconazole in the primary treatment of candidemia was demonstrated in an open, comparative study. Three hundred and seventy (370) non-neutropenic patients with documented candidemia (positive blood culture and clinical signs of infection) were included in the study, of which 248 were treated with voriconazole. The patient population was seriously ill, with approximately 50% of subjects in the intensive care unit and 40% mechanically ventilated at baseline. The median treatment duration was 15 days in both treatment arms. A successful response (resolution/improvement in all clinical signs and symptoms of infection, blood cultures negative for Candida, infected deep tissue sites negative for Candida) was seen in 41% of patients in both treatment arms 12 weeks after the End of Therapy (EOT).
In this analysis, patients who did not have an assessment 12 weeks after EOT were set to failure. According to a secondary analysis, which compared response rates at the latest time point most relevant to the evaluation of the patient (EOT, or 2, 6, or 12 weeks after EOT), voriconazole and the regimen of amphotericin B followed by fluconazole had response rates of 65% and 71%, respectively.
Serious refractory Candida infections:
The study comprised 55 patients with serious refractory systemic Candida infections (including candidemia, disseminated and other invasive candidiasis) where prior antifungal treatment, particularly with fluconazole, had been ineffective. Successful response was seen in 24 patients (15 complete, 9 partial responses). In fluconazole-resistant non-albicans species, a successful outcome was seen in 3/3
C. krusei (complete responses) and 6/8
C. glabrata (5 complete, 1 partial response) infections. The clinical efficacy data were supported by limited susceptibility data.
Other serious rare fungal pathogens: Voriconazole was shown to be effective against the following rare fungal pathogens:
Scedosporium spp.: Successful response to voriconazole therapy was seen in 16 of 28 patients (55%) with
S. apiospermum and in 2 of 7 patients (29%) with
S. prolificans infection. In addition, a successful response was seen in 1 of 3 patients with mixed organism infections.
Fusarium spp.: Seven of 17 (41%) patients were successfully treated with voriconazole. Of these 7 patients, 3 had eye, 1 had sinus, and 3 had disseminated infection. Four additional patients with fusariosis had an infection caused by several organisms; two of them had a successful outcome.
The majority of patients receiving voriconazole treatment of the previously mentioned rare infections were intolerant of, or refractory to, prior antifungal therapy.
Primary Prophylaxis of Invasive Fungal Infections - Efficacy in hematopoietic stem cell transplant (HSCT) recipients without prior proven or probable invasive fungal infection (IFI): Voriconazole was compared to itraconazole as primary prophylaxis in an open-label, comparative, multicenter study of adult and adolescent allogeneic HSCT recipients without prior proven or probable IFI. Success was defined as the ability to continue study drug prophylaxis for 100 days after HSCT (without stopping for >14 days) and survival with no proven or probable IFI for 180 days after HSCT. The modified intent-to-treat (MITT) group included 465 allogeneic HSCT recipients, with myeloablative (58%) or reduced-intensity (42%) conditioning regimens. Prophylaxis with study drug was started immediately after HSCT: 224 received voriconazole and 241 received itraconazole. The median duration of study drug prophylaxis was 96 days for voriconazole and 68 days for itraconazole in the MITT group.
Success rates and other secondary endpoints are presented in the table as follows: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Secondary Prophylaxis of IFI - Efficacy in HSCT recipients with prior proven or probable IFI: Voriconazole was investigated as secondary prophylaxis in an open-label, non-comparative, multicenter study of adult allogeneic HSCT recipients with prior proven or probable IFI. The primary endpoint was the rate of occurrence of proven and probable IFI during the first year after HSCT. The MITT group included 40 patients with prior IFI, including 31 with aspergillosis, 5 with candidiasis, and 4 with other IFI. The median duration of study drug prophylaxis was 95.5 days in the MITT group.
Proven or probable IFIs developed in 7.5% (3/40) of patients during the first year after HSCT, including one candidemia, one scedosporiosis (both relapses of prior IFI), and one zygomycosis. The survival rate at Day 180 was 80.0% (32/40) and at 1 year was 70.0% (28/40).
Duration of Treatment: Intravenous and oral voriconazole allows flexibility in patient care and the possibility of prolonged treatment where indicated. In clinical trials, 714 patients received voriconazole therapy for greater than 12 weeks, with 155 subjects receiving voriconazole for over 6 months.
Clinical Studies in Children: Fifty-three pediatric patients aged 2 to <18 years were treated with voriconazole in two prospective, open-label, non-comparative, multi-center clinical trials. One study enrolled 31 patients with possible, proven or probable invasive aspergillosis (IA), of whom 14 patients had proven or probable IA and were included in the MITT efficacy analyses. The second study enrolled 22 patients with invasive candidiasis including candidaemia (ICC), and esophageal candidiasis (EC) requiring either primary or salvage therapy, of whom 17 were included in the MITT efficacy analyses. Of the total of 31 patients included in the MITT analyses, 14 were 2 to <12 years old (5 patients with IA and 9 with ICC or EC) and 17 were 12 to <18 years old (9 patients with IA and 8 with ICC and EC). The overall rates of global response were 64.3% (9/14) at 6 weeks for patients with IA, 85.7% (6/7) at EOT for patients with ICC and 70% (7/10) at EOT for patients with EC. In subjects with IA, the success rate was 40% (2/5) for patients 2 to <12 years and 77.8% (7/9) for patients 12 to <18 years of age.
Clinical Studies Examining QT Interval: A placebo-controlled, randomized, single-dose, crossover study to evaluate the effect on the QT interval of healthy volunteers was conducted with three oral doses of voriconazole and ketoconazole. The placebo-adjusted mean maximum increases in QTc from baseline after 800, 1,200 and 1,600 mg of voriconazole were 5.1, 4.8, and 8.2 msec, respectively, and 7.0 msec for ketoconazole 800 mg. No subject in any group had an increase in QTc of ≥60 msec from baseline. No subject experienced an interval exceeding the potentially clinically relevant threshold of 500 msec.
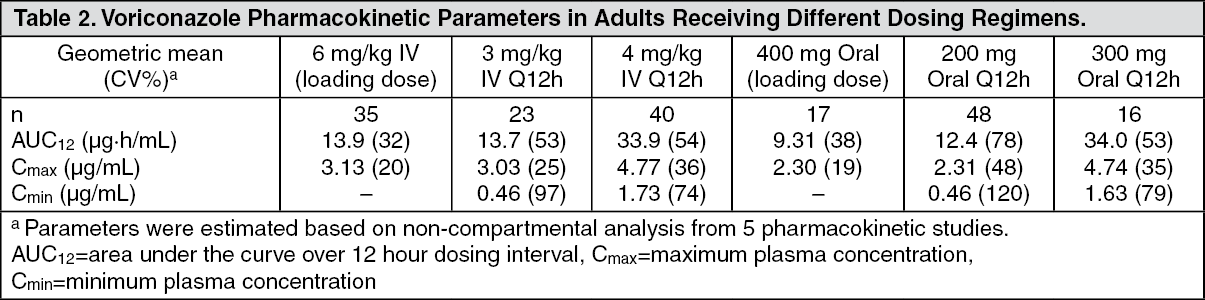
Pharmacokinetics: General Pharmacokinetic Characteristics: The pharmacokinetics of voriconazole have been characterized in healthy subjects, special populations and patients. During oral administration of 200 mg or 300 mg twice daily for 14 days in patients at risk of aspergillosis (mainly patients with malignant neoplasms of lymphatic or hematopoietic tissue), the observed pharmacokinetic characteristics of rapid and consistent absorption, accumulation and non-linear pharmacokinetics were in agreement with those observed in healthy subjects.
The pharmacokinetics of voriconazole are non-linear due to saturation of its metabolism. Greater than proportional increase in exposure is observed with increasing dose. It is estimated that, on average, increasing the oral dose from 200 mg twice daily to 300 mg twice daily leads to an approximately 2.5-fold increase in exposure (AUC
τ). The oral maintenance dose of 200 mg (or 100 mg for patients less than 40 kg) achieves a voriconazole exposure similar to 3 mg/kg IV. A 300 mg (or 150 mg for patients less than 40 kg) oral maintenance dose achieves an exposure similar to 4 mg/kg IV (see table as follows). (See Table 2.)
Click on icon to see table/diagram/image
When the recommended intravenous or oral loading dose regimens are administered, plasma concentrations close to steady state are achieved within the first 24 hours of dosing (e.g., 6 mg/kg IV every 12 hours on day 1 followed by 3 mg/kg IV every 12 hours; 400 mg oral every 12 hours on day 1 followed by 200 mg oral every 12 hours). Without the loading dose, accumulation occurs during twice daily multiple dosing with steady-state plasma voriconazole concentrations being achieved by day 6 in the majority of subjects.
Absorption: Voriconazole is rapidly and almost completely absorbed following oral administration, with maximum plasma concentrations (C
max) achieved 1 to 2 hours after dosing. The oral bioavailability of voriconazole is estimated to be 96%. Bioequivalence was established between the 200 mg tablet and the 40 mg/mL oral suspension when administered as a 400 mg every 12 hours loading dose followed by a 200 mg every 12 hours maintenance dose. When multiple doses of voriconazole are administered with high fat meals, C
max and AUC
τ are reduced by 34% and 24%, respectively, when administered as a tablet and by 58% and 37%, respectively, when administered as the oral suspension.
The absorption of voriconazole is not affected by changes in gastric pH.
Distribution: The volume of distribution at steady state for voriconazole is estimated to be 4.6 L/kg, suggesting extensive distribution into tissues. Plasma protein binding is estimated to be 58%.
Cerebrospinal fluid samples from eight patients in a compassionate programme showed detectable voriconazole concentrations in all patients.
Metabolism: In vitro studies showed that voriconazole is metabolised by the hepatic cytochrome P450 isoenzymes, CYP2C19, CYP2C9 and CYP3A4.
The inter-individual variability of voriconazole pharmacokinetics is high.
In vivo studies indicated that CYP2C19 plays a key role in the metabolism of voriconazole. This enzyme exhibits genetic polymorphism. For example, 15%-20% of Asian populations may be expected to be poor metabolisers. For Caucasians and Blacks, the prevalence of poor metabolisers is 3%-5%. Studies conducted in Caucasian and Japanese healthy subjects have shown that poor metabolisers have, on average, 4-fold higher voriconazole exposure (AUC
τ) than their homozygous extensive metaboliser counterparts. Subjects who are heterozygous extensive metabolisers have on average 2-fold higher voriconazole exposure than their homozygous extensive metaboliser counterparts.
The major metabolite of voriconazole is the N-oxide, which accounts for 72% of the circulating radiolabelled metabolites in plasma. This metabolite has minimal antifungal activity and does not contribute to the overall efficacy of voriconazole.
Excretion: Voriconazole is eliminated via hepatic metabolism with less than 2% of the dose excreted unchanged in the urine.
After administration of a radiolabelled dose of voriconazole, approximately 80% of the radioactivity is recovered in the urine after multiple intravenous dosing and 83% in the urine after multiple oral dosing. The majority (>94%) of the total radioactivity is excreted in the first 96 hours after both oral and intravenous dosing.
The terminal half-life of voriconazole depends on dose and is approximately 6 hours following 200 mg (orally). Because of non-linear pharmacokinetics, the terminal half-life is not useful in the prediction of the accumulation or elimination of voriconazole.
Pharmacokinetics in Special Patient Groups: Gender: In an oral multiple dose study, C
max and AUC
τ for healthy young females were 83% and 113% higher, respectively, than in healthy young males (18-45 years), after tablet dosing. In the same study, no significant differences in C
max and AUC
τ were observed between healthy elderly males and healthy elderly females (≥65 years). In a similar study, after dosing with the oral suspension, the mean AUC for healthy young females was 45% higher than in healthy young males, whereas the mean C
max was comparable between genders. The steady-state trough voriconazole concentrations (C
min) seen in females were 100% and 91% higher than in males receiving the tablet and the oral suspension, respectively.
In the clinical program, no dosage adjustment was made on the basis of gender. The safety profile and plasma concentrations observed in male and female patients were similar. Therefore, no dosage adjustment based on gender is necessary.
Elderly: In an oral multiple dose study C
max and AUC
τ in healthy elderly males (≥65 years) were 61% and 86% higher, respectively, than in healthy young males (18-45 years). No significant differences in C
max and AUC
τ were observed between healthy elderly females (≥65 years) and healthy young females (18-45 years).
In the therapeutic studies, no dosage adjustment was made on the basis of age. A relationship between plasma concentrations and age was observed. However, the safety profile of voriconazole in young and elderly patients was similar and, therefore, no dosage adjustment is necessary for the elderly.
Pediatrics: The recommended doses in children and adolescent patients are based on a population pharmacokinetic analysis of data pooled from 112 immunocompromised pediatric patients aged 2 to <12 years old and 26 immunocompromised adolescent patients aged 12 to ≤17 years. Multiple intravenous doses of 3, 4, 6, 7 and 8 mg/kg twice daily and multiple oral doses (using the powder for oral suspension) of 4 mg/kg, 6 mg/kg and 200 mg twice daily were evaluated in 3 pediatric pharmacokinetic studies. Intravenous loading doses of 6 mg/kg IV twice daily on day 1 followed by 4 mg/kg intravenous dose twice daily and 300 mg oral tablets twice daily were evaluated in one adolescent pharmacokinetic study. Larger inter-subject variability was observed in pediatric patients compared to adults.
A comparison of the pediatric and adult population pharmacokinetic data indicated that the predicted total exposure (AUC
τ) in children following administration of a 9 mg/kg IV loading dose was comparable to that in adults following a 6 mg/kg IV loading dose. The predicted total exposures in children following IV maintenance doses of 4 and 8 mg/kg twice daily were comparable to those in adults following 3 and 4 mg/kg IV twice daily, respectively. The predicted total exposure in children following an oral maintenance dose of 9 mg/kg (maximum of 350 mg) twice daily was comparable to that in adults following 200 mg oral twice daily. An 8 mg/kg intravenous dose will provide voriconazole exposure approximately 2-fold higher than a 9 mg/kg oral dose.
The higher intravenous maintenance dose in pediatric patients relative to adults reflects the higher elimination capacity in pediatric patients due to a greater liver mass to body mass ratio.
Oral bioavailability may however, be limited in pediatric patients with malabsorption and very low body weight for their age. In that case, intravenous voriconazole administration is recommended.
Voriconazole exposures in the majority of adolescent patients were comparable to those in adults receiving the same dosing regimens. However, lower voriconazole exposure was observed in some young adolescents with low body weight compared to adults. It is likely that these subjects may metabolise voriconazole more similarly to children than to adults. Based on the population pharmacokinetic analysis, 12- to 14-year-old adolescents weighing less than 50 kg should receive children's doses (see Dosage & Administration).
Renal Impairment: In a single oral dose (200 mg) study in subjects with normal renal function and mild (creatinine clearance 41-60 mL/min) to severe (creatinine clearance <20 mL/min) renal impairment, the pharmacokinetics of voriconazole were not significantly affected by renal impairment. The plasma protein binding of voriconazole was similar in subjects with different degrees of renal impairment. See dosing and monitoring recommendations under Dosage & Administration and Precautions.
In patients with moderate to severe renal dysfunction (serum creatinine levels ≥220 micromol/L (2.5 mg/dL), accumulation of the intravenous vehicle, SBECD, occurs. See dosing and monitoring recommendations under Dosage & Administration and Precautions.
Hepatic Impairment: After a single oral dose (200 mg), AUC was 233% higher in subjects with mild to moderate hepatic cirrhosis (Child-Pugh A and B) compared with subjects with normal hepatic function. Protein binding of voriconazole was not affected by impaired hepatic function.
In a multiple oral dose study, AUC
τ was similar in subjects with moderate hepatic cirrhosis (Child-Pugh B) given maintenance doses of 100 mg twice daily and subjects with normal hepatic function given 200 mg twice daily. No pharmacokinetic data are available for patients with severe hepatic cirrhosis (Child-Pugh C). For dosing information, refer to use in patients with hepatic impairment (see Dosage & Administration).
Toxicology: Preclinical safety data: Repeated-dose toxicity studies with voriconazole indicated the liver to be the target organ. Hepatotoxicity occurred at plasma exposures similar to those obtained at therapeutic doses in humans, in common with other antifungal agents. In rats, mice and dogs, voriconazole also induced minimal adrenal changes. Conventional studies of safety pharmacology, genotoxicity or carcinogenic potential did not reveal a special hazard for humans.
In reproduction studies, voriconazole was shown to be teratogenic in rats and embryotoxic in rabbits at systemic exposures equal to those obtained in humans with therapeutic doses. In the pre- and post-natal development study in rats at exposures lower than those obtained in humans with therapeutic doses, voriconazole prolonged the duration of gestation and labor and produced dystocia with consequent maternal mortality and reduced perinatal survival of pups. The effects on parturition are probably mediated by species-specific mechanisms, involving reduction of estradiol levels, and are consistent with those observed with other azole antifungal agents. Voriconazole administration induced no impairment of male or female fertility in rats at exposures similar to those obtained in humans at therapeutic doses.
Preclinical data on the intravenous vehicle, SBECD indicated that the main effects were vacuolation of urinary tract epithelium and activation of macrophages in the liver and lungs in the repeated-dose toxicity studies.
Microbiology: In vitro, voriconazole displays broad-spectrum antifungal activity with antifungal potency against
Candida species (including fluconazole resistant
C. krusei and resistant strains of
C. glabrata and
C. albicans) and fungicidal activity against all
Aspergillus species tested. In addition, voriconazole shows
in vitro fungicidal activity against emerging fungal pathogens, including those such as Scedosporium
or Fusarium which have limited susceptibility to existing antifungal agents.
Clinical efficacy (with partial or complete response, see Clinical Experience as previously mentioned) has been demonstrated for
Aspergillus spp. including
A. flavus,
A. fumigatus,
A. terreus,
A. niger,
A. nidulans,
Candida spp., including
C. albicans,
C. glabrata,
C. krusei,
C. parapsilosis and
C. tropicalis and limited numbers of
C. dubliniensis,
C. inconspicua and
C. guilliermondii, Scedosporium spp., including
S. apiospermum,
S. prolificans and
Fusarium spp.
Other treated fungal infections (with often partial or complete response) included isolated cases of
Alternaria spp.,
Blastomyces dermatitidis,
Blastoschizomyces capitatus,
Cladosporium spp.,
Coccidioides immitis,
Conidiobolus coronatus,
Cryptococcus neoformans,
Exserohilum rostratum,
Exophiala spinifera,
Fonsecaea pedrosoi,
Madurella mycetomatis,
Paecilomyces lilacinus,
Penicillium spp. including
P. marneffei,
Phialophora richardsiae,
Scopulariopsis brevicaulis and
Trichosporon spp. including
T. beigelii infections.
In vitro activity against clinical isolates has been observed for
Acremonium spp.,
Alternaria spp.,
Bipolaris spp.,
Cladophialophora spp.,
Histoplasma capsulatum, with most strains being inhibited by concentrations of voriconazole in the range 0.05 to 2 mcg/mL.
In vitro activity against the following pathogens has been shown, but the clinical significance is unknown:
Curvularia spp. and
Sporothrix spp.
Breakpoints: Specimens for fungal culture and other relevant laboratory studies (serology, histopathology) should be obtained prior to therapy to isolate and identify causative organisms. Therapy may be instituted before the results of the cultures and other laboratory studies are known; however, once these results become available, anti-infective therapy should be adjusted accordingly.
The species most frequently involved in causing human infections include
C. albicans,
C. parapsilosis,
C. tropicalis,
C. glabrata, and
C. krusei, all of which usually exhibit minimum inhibitory concentrations (MICs) of less than 1 mg/L for voriconazole.
However, the
in vitro activity of voriconazole against
Candida species is not uniform. Specifically, for
C. glabrata, the MICs of voriconazole for fluconazole-resistant isolates are proportionally higher than are those of fluconazole-susceptible isolates. Therefore, every attempt should be made to identify Candida to species level. If antifungal susceptibility testing available, the MIC results may be interpreted using breakpoint criteria.
European Committee on Antimicrobial Susceptibility Testing (EUCAST) Breakpoints: Candida species: The interpretive standards for voriconazole against
Candida species are applicable only to tests performed using EUCAST microbroth dilution reference method for minimum inhibitory concentrations (MICs) read at 24 hours.
(See Table 3.)
Click on icon to see table/diagram/image
Clinical and Laboratory Standards Institute (CLSI) Breakpoints: Breakpoint criteria established by CLSI: Susceptibility Testing Methods:
Aspergillus species and other filamentous fungi: No interpretive criteria have been established for
Aspergillus species and other filamentous fungi.
Candida species: The interpretive standards for voriconazole against
Candida species are applicable only to tests performed using Clinical and Laboratory Standards Institute (CLSI) microbroth dilution reference method M27 for MIC read at 48 hours or disk diffusion reference method M44 for zone diameter read at 24 hours.
Broth Dilution Techniques: Quantitative methods are used to determine antifungal MICs. These MICs provide estimates of the susceptibility of
Candida species to antifungal agents.
MICs should be determined using a standardized procedure at 48 hours. Standardized procedures are based on a microdilution method (broth) with standardized inoculums concentrations and standardized concentrations of voriconazole powder. The MIC values should be interpreted according to the criteria provided in the table as follows.
Diffusion Techniques: Qualitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of
Candida species to an antifungal agent.
One such standardized procedure requires the use of standardized inoculum concentrations.
This procedure uses paper discs impregnated with 1 microgram of voriconazole to test the susceptibility of yeasts to voriconazole. Disc diffusion interpretive criteria are also provided in the table as follows. (See Table 4.)
Click on icon to see table/diagram/image
The susceptible category implies that isolates are inhibited by the usually achievable concentrations of antifungal agent tested when the recommended dosage is used for the site of infection. The susceptible-dose dependent category implies that an infection due to the isolate may be appropriately treated in body sites where the drugs are physiologically concentrated or when a high dosage of drug is used. The resistant category implies that isolates are not inhibited by the usually achievable concentrations of the agent with normal dosage schedules and clinical efficacy of the agent against the isolate has not been reliably shown in treatment studies.
Quality Control: Standardized susceptibility test procedures require the use of quality control organisms to control the technical aspects of the test procedures. Standard voriconazole powder and 1 μg discs should provide the following range of values noted in the table as follows.
NOTE: Quality control microorganisms are specific strains of organisms with intrinsic biological properties relating to resistance mechanisms and their genetic expression within fungi; the specific strains used for microbiological control are not clinically significant. (See Table 5.)
Click on icon to see table/diagram/image




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image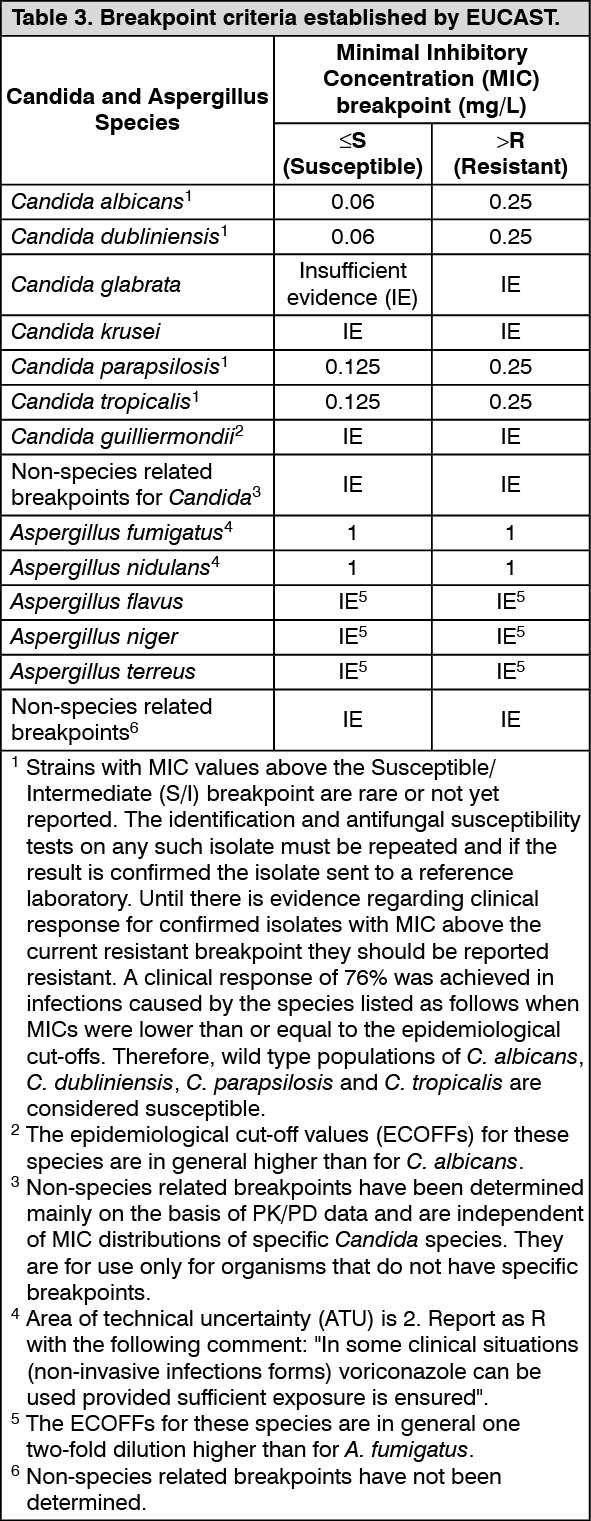 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image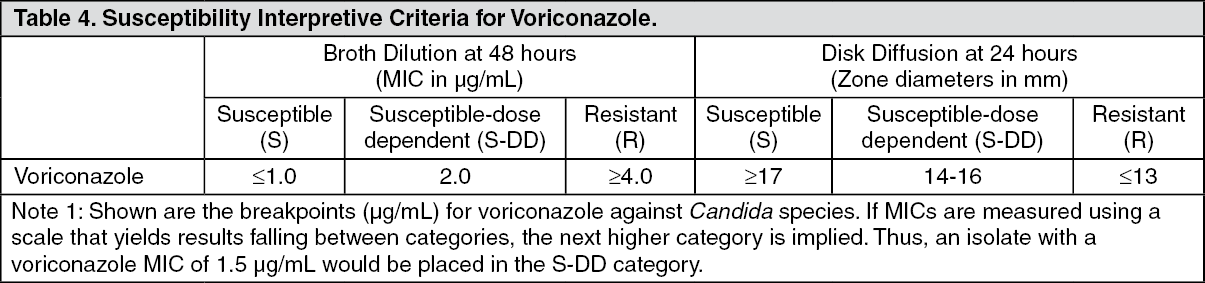 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image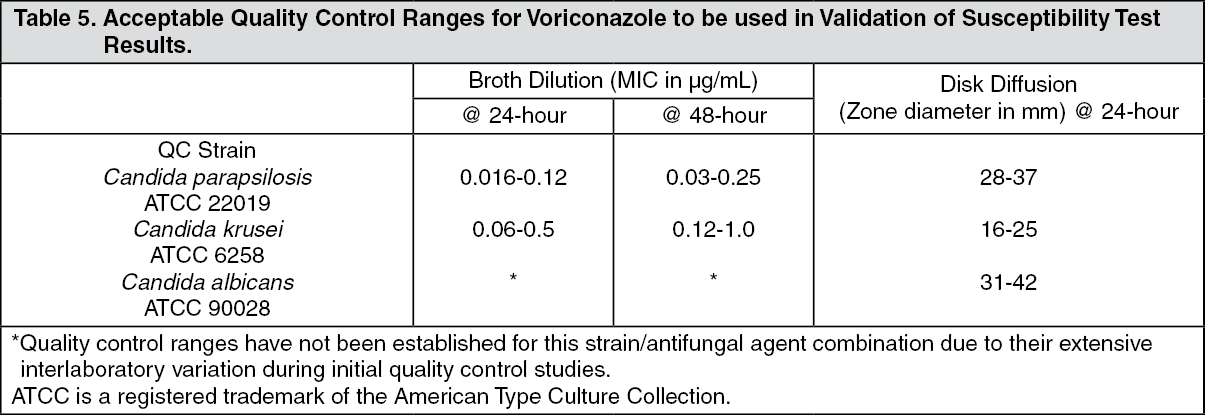 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image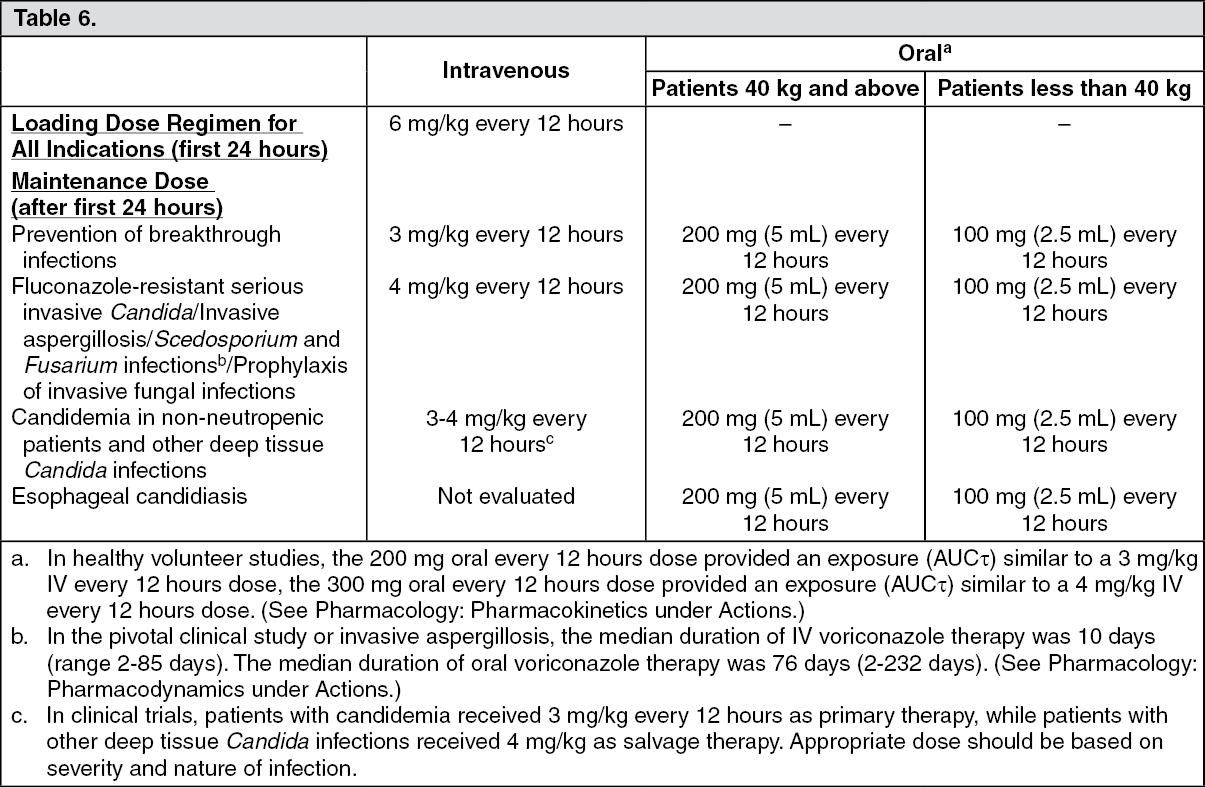 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image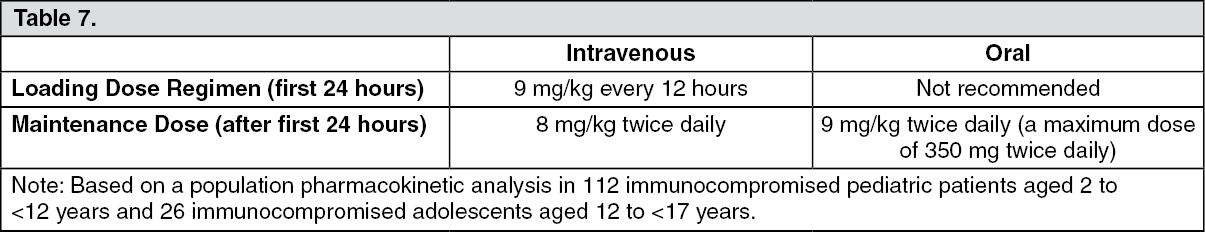 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 200 mg4d6ada60-9d46-41c1-944a-aa99008d809b.GIF)

 Sign Out
Sign Out
