Increased Mortality in Elderly Patients with Dementia-Related Psychosis: Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of 17 placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6- to 1.7-times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. LATUDA is not approved for the treatment of patients with dementia-related psychosis [see Precautions].
Suicidal Thoughts and Behaviors in Pediatric and Young Adult Patients: In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients, and over 4,400 pediatric patients, the incidence of suicidal thoughts and behaviors in pediatric and young adult patients was greater in antidepressant-treated patients than in placebo-treated patients. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 5.
No suicides occurred in any of the pediatric studies. There were suicides in the adult studies, but the number was not sufficient to reach any conclusion about antidepressant drug effect on suicide. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
It is unknown whether the risk of suicidal thoughts and behaviors in pediatric and young adult patients extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with MDD that antidepressants delay the recurrence of depression.
Monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing LATUDA, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts or behaviors.
Cerebrovascular Adverse Reactions, Including Stroke in Elderly Patients with Dementia-Related Psychosis: In placebo-controlled trials with risperidone, aripiprazole, and olanzapine in elderly subjects with dementia, there was a higher incidence of cerebrovascular adverse reactions (cerebrovascular accidents and transient ischemic attacks), including fatalities, compared to placebo-treated subjects. LATUDA is not approved for the treatment of patients with dementia-related psychosis [see Precautions].
Neuroleptic Malignant Syndrome: A potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) has been reported in association with administration of antipsychotic drugs, including LATUDA. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability. Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure.
If NMS is suspected, immediately discontinue LATUDA and provide intensive symptomatic treatment and monitoring.
Tardive Dyskinesia: Tardive dyskinesia is a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements that can develop in patients treated with antipsychotic drugs. Although the prevalence of the syndrome appears to be highest among the elderly, especially elderly women, it is impossible to rely upon prevalence estimates to predict, at the inception of antipsychotic treatment, which patients are likely to develop the syndrome. Whether antipsychotic drug products differ in their potential to cause tardive dyskinesia is unknown.
The risk of developing tardive dyskinesia and the likelihood that it will become irreversible are believed to increase as the duration of treatment and the total cumulative dose of antipsychotic drugs administered to the patient increase. However, the syndrome can develop, although much less commonly, after relatively brief treatment periods at low doses or may even arise after discontinuation of treatment.
The syndrome may remit, partially or completely, if antipsychotic treatment is withdrawn. Antipsychotic treatment, itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome and thereby may possibly mask the underlying process. The effect that symptomatic suppression has upon the long-term course of the syndrome is unknown.
Given these considerations, LATUDA should be prescribed in a manner that is most likely to minimize the occurrence of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients who suffer from a chronic illness that (1) is known to respond to antipsychotic drugs, and (2) for whom alternative, equally effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, the smallest dose and the shortest duration of treatment producing a satisfactory clinical response should be sought. The need for continued treatment should be reassessed periodically.
If signs and symptoms of tardive dyskinesia appear in a patient on LATUDA, drug discontinuation should be considered. However, some patients may require treatment with LATUDA despite the presence of the syndrome.
Metabolic Changes: Atypical antipsychotic drugs have been associated with metabolic changes that may increase cardiovascular/cerebrovascular risk. These metabolic changes include hyperglycemia, dyslipidemia, and body weight gain. While all of the drugs in the class have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia and Diabetes Mellitus: Hyperglycemia, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, has been reported in patients treated with atypical antipsychotics. Assessment of the relationship between atypical antipsychotic use and glucose abnormalities is complicated by the possibility of an increased background risk of diabetes mellitus in patients with schizophrenia and the increasing incidence of diabetes mellitus in the general population. Given these confounders, the relationship between atypical antipsychotic use and hyperglycemia-related adverse events is not completely understood. However, epidemiological studies suggest an increased risk of hyperglycemia-related adverse events in patients treated with the atypical antipsychotics. Precise risk estimates for hyperglycaemia-related adverse events in patients treated with atypical antipsychotics are not available.
Patients with an established diagnosis of diabetes mellitus who are started on atypical antipsychotics should be monitored regularly for worsening of glucose control. Patients with risk factors for diabetes mellitus (e.g., obesity, family history of diabetes) who are starting treatment with atypical antipsychotics should undergo fasting blood glucose testing at the beginning of treatment and periodically during treatment. Any patient treated with atypical antipsychotics should be monitored for symptoms of hyperglycemia including polydipsia, polyuria, polyphagia, and weakness. Patients who develop symptoms of hyperglycemia during treatment with atypical antipsychotics should undergo fasting blood glucose testing. In some cases, hyperglycemia has resolved when the atypical antipsychotic was discontinued; however, some patients required continuation of anti-diabetic treatment despite discontinuation of the suspect drug.
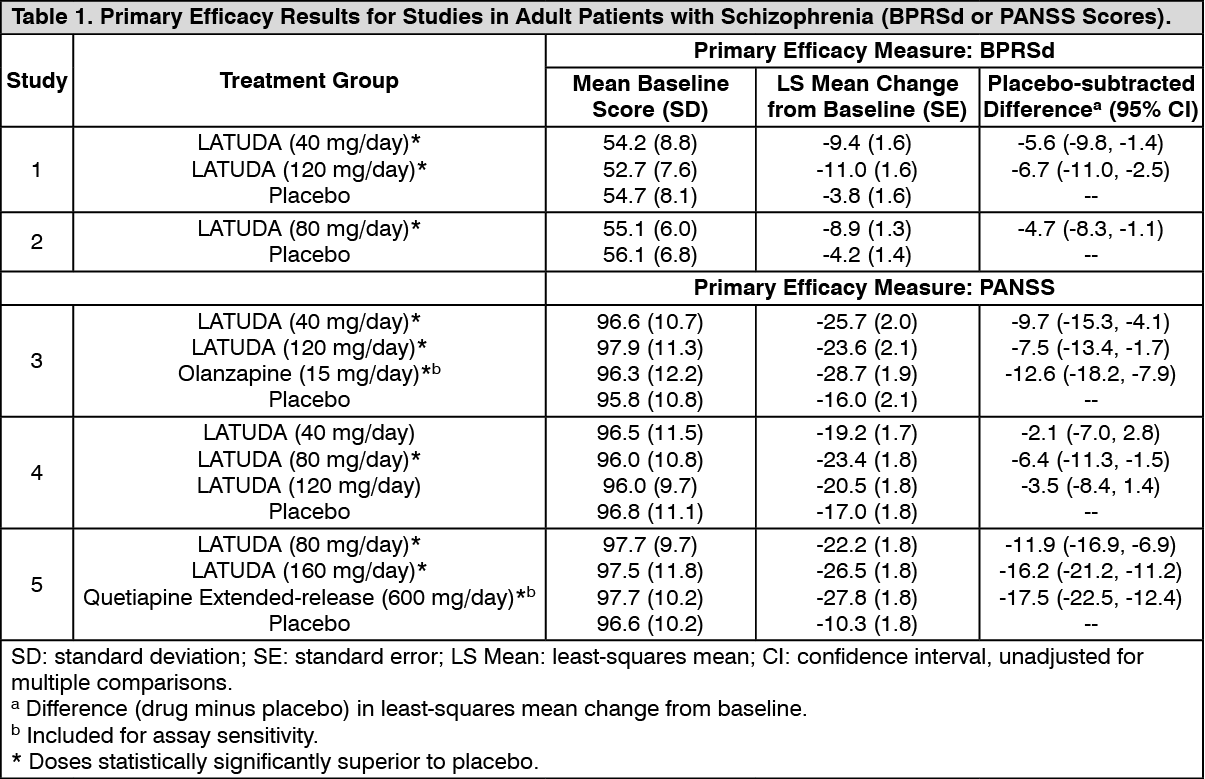
Schizophrenia: Adults: Pooled data from short-term, placebo-controlled schizophrenia studies are presented in Table 6. (See Table 6.)
Click on icon to see table/diagram/image
In the uncontrolled, longer-term schizophrenia studies (primarily open-label extension studies), LATUDA was associated with a mean change in glucose of +1.8 mg/dL at week 24 (n=355), +0.8 mg/dL at week 36 (n=299) and +2.3 mg/dL at week 52 (n=307).
Adolescents: In studies of adolescents and adults with schizophrenia, changes in fasting glucose were similar. In the short-term, placebo-controlled study of adolescents, fasting serum glucose mean values were -1.3 mg/dL for placebo (n=95), +0.1 mg/dL for 40 mg/day (n=90), and +1.8 mg/dL for 80 mg/day (n=92).
Bipolar Depression: Adults: Monotherapy: Data from the adult short-term, flexible-dose, placebo-controlled monotherapy bipolar depression study are presented in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who received LATUDA as monotherapy in the short-term study and continued in the longer-term study, had a mean change in glucose of +1.2 mg/dL at week 24 (n=129).
Adjunctive Therapy with Lithium or Valproate: Data from the adult short-term, flexible-dosed, placebo-controlled adjunctive therapy bipolar depression studies are presented in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who received LATUDA as adjunctive therapy with either lithium or valproate in the short-term study and continued in the longer-term study, had a mean change in glucose of +1.7 mg/dL at week 24 (n=88).
Pediatric Patients (10 to 17 years): In studies of pediatric patients 10 to 17 years and adults with bipolar depression, changes in fasting glucose were similar. In the 6-week, placebo-controlled study of pediatric patients with bipolar depression, mean change in fasting glucose was +1.6 mg/dL for LATUDA 20 to 80 mg/day (n=145) and -0.5 mg/dL for placebo (n=145).
Pediatric Patients (6 to 17 years): In a 104-week, open-label study in pediatric patients with schizophrenia, bipolar depression, or autistic disorder, 7 % of patients with a normal baseline fasting glucose experienced a shift to high at endpoint while taking lurasidone.
Dyslipidemia: Undesirable alterations in lipids have been observed in patients treated with atypical antipsychotics.
Schizophrenia: Adults: Pooled data from short-term, placebo-controlled schizophrenia studies are presented in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
In the uncontrolled, longer-term schizophrenia studies (primarily open-label extension studies), LATUDA was associated with a mean change in total cholesterol and triglycerides of -3.8 (n=356) and -15.1 (n=357) mg/dL at week 24, -3.1 (n=303) and -4.8 (n=303) mg/dL at week 36 and -2.5 (n=307) and -6.9 (n=307) mg/dL at week 52, respectively.
Adolescents: In the adolescent short-term, placebo-controlled study, fasting serum cholesterol mean values were -9.6 mg/dL for placebo (n=95), -4.4 mg/dL for 40 mg/day (n=89), and +1.6 mg/dL for 80 mg/day (n=92), and fasting serum triglyceride mean values were +0.1 mg/dL for placebo (n=95), -0.6 mg/dL for 40 mg/day (n=89), and +8.5 mg/dL for 80 mg/day (n=92).
Bipolar Depression: Adults: Monotherapy: Data from the adult short-term, flexible-dosed, placebo-controlled, monotherapy bipolar depression study are presented in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who received LATUDA as monotherapy in the short-term and continued in the longer-term study had a mean change in total cholesterol and triglycerides of -0.5 mg/dL (n=130) and -1.0 mg/dL (n=130) at week 24, respectively.
Adjunctive Therapy with Lithium or Valproate: Data from the adult short-term, flexible-dosed, placebo-controlled, adjunctive therapy bipolar depression studies are presented in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who received LATUDA, as adjunctive therapy with either lithium or valproate in the short-term study and continued in the longer-term study, had a mean change in total cholesterol and triglycerides of -0.9 (n=88) and +5.3 (n=88) mg/dL at week 24, respectively.
Pediatric Patients (10 to 17 years): In the 6-week, placebo-controlled bipolar depression study with pediatric patients 10 to 17 years, mean change in fasting cholesterol was -6.3 mg/dL for LATUDA 20 to 80 mg/day (n=144) and -1.4 mg/dL for placebo (n=145), and mean change in fasting triglyceride was -7.6 mg/dL for LATUDA 20 to 80 mg/day (n=144) and +5.9 mg/dL for placebo (n=145).
Pediatric Patients (6 to 17 years): In a 104-week, open-label study of pediatric patients with schizophrenia, bipolar depression, or autistic disorder, shifts in baseline fasting cholesterol from normal to high at endpoint were reported in 12% (total cholesterol), 3% (LDL cholesterol), and shifts in baseline from normal to low were reported in 27% (HDL cholesterol) of patients taking lurasidone. Of patients with normal baseline fasting triglycerides, 12% experienced shifts to high.
Weight Gain: Weight gain has been observed with atypical antipsychotic use. Clinical monitoring of weight is recommended.
Schizophrenia: Adults: Pooled data from short-term, placebo-controlled schizophrenia studies are presented in Table 12. The mean weight gain was +0.43 kg for LATUDA-treated patients compared to -0.02 kg for placebo-treated patients. Change in weight from baseline for olanzapine was +4.15 kg and for quetiapine extended-release was +2.09 kg in Studies 3 and 5 [see Pharmacology: Pharmacodynamics: Clinical Studies under Actions], respectively. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 4.8% for LATUDA-treated patients and 3.3% for placebo-treated patients. (See Table 12.)
Click on icon to see table/diagram/image
In the uncontrolled, longer-term schizophrenia studies (primarily open-label extension studies), LATUDA was associated with a mean change in weight of -0.69 kg at week 24 (n=755), -0.59 kg at week 36 (n=443) and -0.73 kg at week 52 (n=377).
Adolescents: Data from the short-term, placebo-controlled adolescent schizophrenia study are presented in Table 13. The mean change in weight gain was +0.5 kg for LATUDA-treated patients compared to +0.2 kg for placebo-treated patients. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 3.3% for LATUDA-treated patients and 4.5% for placebo-treated patients. (See Table 13.)
Click on icon to see table/diagram/image
Bipolar Depression: Adults: Monotherapy: Data from the adult short-term, flexible-dosed, placebo-controlled monotherapy bipolar depression study are presented in Table 14. The mean change in weight gain was +0.29 kg for LATUDA-treated patients compared to -0.04 kg for placebo-treated patients. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 2.4% for LATUDA-treated patients and 0.7% for placebo-treated patients. (See Table 14.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who received LATUDA as monotherapy in the short-term and continued in the longer-term study had a mean change in weight of -0.02 kg at week 24 (n=130).
Adjunctive Therapy with Lithium or Valproate: Data from the adult short-term, flexible-dosed, placebo-controlled adjunctive therapy bipolar depression studies are presented in Table 15. The mean change in weight gain was +0.11 kg for LATUDA-treated patients compared to +0.16 kg for placebo-treated patients. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 3.1% for LATUDA-treated patients and 0.3% for placebo-treated patients. (See Table 15.)
Click on icon to see table/diagram/image
In the uncontrolled, open-label, longer-term bipolar depression study, patients who were treated with LATUDA, as adjunctive therapy with either lithium or valproate in the short-term and continued in the longer-term study, had a mean change in weight of +1.28 kg at week 24 (n=86).
Pediatric Patients (10 to 17 years): Data from the 6-week, placebo-controlled bipolar depression study in patients 10 to 17 years are presented in Table 16. The mean change in weight gain was +0.7 kg for LATUDA-treated patients compared to +0.5 kg for placebo-treated patients. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 4.0% for LATUDA-treated patients and 5.3% for placebo-treated patients. (See Table 16.)
Click on icon to see table/diagram/image
Pediatric Patients (6 to 17 years): In a long-term, open-label study that enrolled pediatric patients with schizophrenia, bipolar depression, or autistic disorder from three short-term, placebo-controlled trials, 54% (378/701) received lurasidone for 104 weeks. The mean increase in weight from open-label baseline to Week 104 was 5.85 kg. To adjust for normal growth, z-scores were derived (measured in standard deviations [SD]), which normalize for the natural growth of children and adolescents by comparisons to age- and sex-matched population standards. A z-score change <0.5 SD is considered not clinically significant. In this trial, the mean change in z-score from open-label baseline to Week 104 was -0.06 SD for body weight and -0.13 SD for body mass index (BMI), indicating minimal deviation from the normal curve for weight gain.
Hyperprolactinemia: As with other drugs that antagonize dopamine D
2 receptors, LATUDA elevates prolactin levels.
Hyperprolactinemia may suppress hypothalamic GnRH, resulting in reduced pituitary gonadotrophin secretion. This, in turn, may inhibit reproductive function by impairing gonadal steroidogenesis in both female and male patients. Galactorrhea, amenorrhea, gynecomastia, and impotence have been reported with prolactin-elevating compounds. Long-standing hyperprolactinemia, when associated with hypogonadism, may lead to decreased bone density in both female and male patients [see Adverse Reactions].
Tissue culture experiments indicate that approximately one-third of human breast cancers are prolactin-dependent
in vitro, a factor of potential importance if the prescription of these drugs is considered in a patient with previously detected breast cancer. As is common with compounds which increase prolactin release, an increase in mammary gland neoplasia was observed in a carcinogenicity study conducted with lurasidone in rats and mice. Neither clinical studies nor epidemiologic studies conducted to date have shown an association between chronic administration of this class of drugs and tumorigenesis in humans, but the available evidence is too limited to be conclusive.
Schizophrenia: Adults: In short-term, placebo-controlled schizophrenia studies, the median change from baseline to endpoint in prolactin levels for LATUDA-treated patients was +0.4 ng/mL and was -1.9 ng/mL in the placebo-treated patients. The median change from baseline to endpoint for males was +0.5 ng/mL and for females was -0.2 ng/mL. Median changes for prolactin by dose are shown in Table 17. (See Table 17.)
Click on icon to see table/diagram/image
The proportion of patients with prolactin elevations ≥5× upper limit of normal (ULN) was 2.8% for LATUDA-treated patients and = 1.0% for placebo-treated patients. The proportion of female patients with prolactin elevations ≥5x ULN was 5.7% for LATUDA-treated patients and = 2.0% for placebo-treated female patients. The proportion of male patients with prolactin elevations ≥5x ULN was 1.6% and 0.6% for placebo-treated male patients.
In the uncontrolled longer-term schizophrenia studies (primarily open-label extension studies), LATUDA was associated with a median change in prolactin of -0.9 ng/mL at week 24 (n=357), -5.3ng/mL at week 36 (n=190) and -2.2 ng/mL at week 52 (n=307).
Adolescents: In the short-term, placebo-controlled adolescent schizophrenia study, the median change from baseline to endpoint in prolactin levels for LATUDA-treated patients was +1.1 ng/mL and was +0.1 ng/mL for placebo-treated patients. For LATUDA-treated patients, the median change from baseline to endpoint for males was +1.0 ng/mL and for females was +2.6 ng/mL. Median changes for prolactin by dose are shown in Table 18. (See Table 18.)
Click on icon to see table/diagram/image
The proportion of patients with prolactin elevations ≥5x ULN was 0.5% for LATUDA-treated patients and 1.0% for placebo-treated patients. The proportion of female patients with prolactin elevations ≥5x ULN was 1.3% for LATUDA-treated patients and 0% for placebo-treated female patients. The proportion of male patients with prolactin elevations ≥5x ULN was 0% for LATUDA treated patients and 1.6% for placebo-treated male patients.
Bipolar Depression: Adults: Monotherapy: The median change from baseline to endpoint in prolactin levels, in the adult short-term, flexible-dosed, placebo-controlled monotherapy bipolar depression study, was +1.7 ng/mL and +3.5 ng/mL with LATUDA 20 to 60 mg/day and 80 to 120 mg/day, respectively compared to +0.3 ng/mL with placebo-treated patients. The median change from baseline to endpoint for males was +1.5 ng/mL and for females was +3.1 ng/mL. Median changes for prolactin by dose range are shown in Table 19. (See Table 19.)
Click on icon to see table/diagram/image
The proportion of patients with prolactin elevations ≥5x upper limit of normal (ULN) was 0.4% for LATUDA-treated patients and 0.0% for placebo-treated patients. The proportion of female patients with prolactin elevations ≥5x ULN was 0.6% for LATUDA-treated patients and 0% for placebo-treated female patients. The proportion of male patients with prolactin elevations ≥5x ULN was 0% and 0% for placebo-treated male patients.
In the uncontrolled, open-label, longer-term bipolar depression study, patients who were treated with LATUDA as monotherapy in the short-term and continued in the longer-term study, had a median change in prolactin of -1.15 ng/mL at week 24 (n=130).
Adjunctive Therapy with Lithium or Valproate: The median change from baseline to endpoint in prolactin levels, in the adult short-term, flexible-dosed, placebo-controlled adjunctive therapy bipolar depression studies was +2.8 ng/mL with LATUDA 20 to 120 mg/day compared to 0.0 ng/mL with placebo-treated patients. The median change from baseline to endpoint for males was +2.4 ng/mL and for females was +3.2 ng/mL. Median changes for prolactin across the dose range are shown in Table 20. (See Table 20.)
Click on icon to see table/diagram/image
The proportion of patients with prolactin elevations ≥5x upper limit of normal (ULN) was 0.0% for LATUDA-treated patients and 0.0% for placebo-treated patients. The proportion of female patients with prolactin elevations ≥5x ULN was 0% for LATUDA-treated patients and 0% for placebo-treated female patients. The proportion of male patients with prolactin elevations ≥5x ULN was 0% and 0% for placebo-treated male patients.
In the uncontrolled, open-label, longer-term bipolar depression study, patients who were treated with LATUDA, as adjunctive therapy with either lithium or valproate, in the short-term and continued in the longer-term study, had a median change in prolactin of -2.9 ng/mL at week 24 (n=88).
Pediatric Patients (10 to 17 years): In the 6-week, placebo-controlled bipolar depression study with pediatric patients 10 to 17 years, the median change from baseline to endpoint in prolactin levels for LATUDA-treated patients was +1.10 ng/mL and was +0.50 ng/mL for placebo-treated patients. For LATUDA-treated patients, the median change from baseline to endpoint for males was +0.85 ng/mL and for females was +2.50 ng/mL. Median changes for prolactin are shown in Table 21. (See Table 21.)
Click on icon to see table/diagram/image
The proportion of patients with prolactin elevations ≥5x ULN was 0% for LATUDA-treated patients and 0.6% for placebo-treated patients. The proportion of female patients with prolactin elevations ≥5x ULN was 0% for LATUDA-treated patients and 1.3% for placebo-treated female patients. No male patients in the placebo or LATUDA treatment groups had prolactin elevations ≥5x ULN.
Pediatric Patients (6 to 17 years): In a 104-week, open-label study of pediatric patients with schizophrenia, bipolar depression, or autistic disorder, the median changes from baseline to endpoint in serum prolactin levels were -0.20 ng/mL (all patients), -0.30 ng/mL (females), and -0.05 ng/mL (males). The proportions of patients with a markedly high prolactin level (≥5 times the upper limit of normal) at any time during open-label treatment were 2% (all patients), 3% (females), and 1% (males).
Adverse events among females in this trial that are potentially prolactin-related include galactorrhea (0.6%). Among male patients in this study, decreased libido was reported in one patient (0.2%) and there were no reports of impotence, gynecomastia, or galactorrhea.
Leukopenia, Neutropenia and Agranulocytosis: Leukopenia/neutropenia has been reported during treatment with antipsychotic agents. Agranulocytosis (including fatal cases) has been reported with other agents in the class.
Possible risk factors for leukopenia/neutropenia include pre-existing low white blood cell count (WBC) and history of drug-induced leukopenia/neutropenia. Patients with a pre-existing low WBC or a history of drug-induced leukopenia/neutropenia should have their complete blood count (CBC) monitored frequently during the first few months of therapy and LATUDA should be discontinued at the first sign of decline in WBC, in the absence of other causative factors.
Patients with neutropenia should be carefully monitored for fever or other symptoms or signs of infection and treated promptly if such symptoms or signs occur. Patients with severe neutropenia (absolute neutrophil count < 1000/mm
3) should discontinue LATUDA and have their WBC followed until recovery.
Orthostatic Hypotension and Syncope: LATUDA may cause orthostatic hypotension and syncope, perhaps due to its α1-adrenergic receptor antagonism. Associated adverse reactions can include dizziness, lightheadedness, tachycardia, and bradycardia. Generally, these risks are greatest at the beginning of treatment and during dose escalation. Patients at increased risk of these adverse reactions or at increased risk of developing complications from hypotension include those with dehydration, hypovolemia, treatment with antihypertensive medication, history of cardiovascular disease (e.g., heart failure, myocardial infarction, ischemia, or conduction abnormalities), history of cerebrovascular disease, as well as patients who are antipsychotic-naïve. In such patients, consider using a lower starting dose and slower titration, and monitor orthostatic vital signs.
Orthostatic hypotension, as assessed by vital sign measurement, was defined by the following vital sign changes: ≥ 20 mm Hg decrease in systolic blood pressure and ≥10 bpm increase in pulse from sitting to standing or supine to standing position.
Schizophrenia: Adults: The incidence of orthostatic hypotension and syncope reported as adverse events from short-term, placebo-controlled schizophrenia studies was (LATUDA incidence, placebo incidence): orthostatic hypotension [0.3% (5/1508), 0.1% (1/708)] and syncope [0.1% (2/1508), 0% (0/708)].
In short-term schizophrenia clinical studies, orthostatic hypotension, as assessed by vital signs, occurred with a frequency of 0.8% with LATUDA 40 mg, 2.1% with LATUDA 80 mg, 1.7% with LATUDA 120 mg and 0.8% with LATUDA 160 mg compared to 0.7% with placebo.
Adolescents: The incidence of orthostatic hypotension reported as adverse events from the short-term, placebo-controlled adolescent schizophrenia study was 0.5% (1/214) in LATUDA-treated patients and 0% (0/112) in placebo-treated patients. No syncope event was reported.
Orthostatic hypotension, as assessed by vital signs, occurred with a frequency of 0% with LATUDA 40 mg and 2.9% with LATUDA 80 mg, compared to 1.8% with placebo.
Bipolar Depression: Adults: Monotherapy: In the adult short-term, flexible-dose, placebo-controlled monotherapy bipolar depression study, there were no reported adverse events of orthostatic hypotension and syncope.
Orthostatic hypotension, as assessed by vital signs, occurred with a frequency of 0.6% with LATUDA 20 to 60 mg and 0.6% with LATUDA 80 to 120 mg compared to 0% with placebo.
Adjunctive Therapy with Lithium or Valproate: In the adult short-term, flexible-dose, placebo-controlled adjunctive therapy bipolar depression therapy studies, there were no reported adverse events of orthostatic hypotension and syncope. Orthostatic hypotension, as assessed by vital signs, occurred with a frequency of 1.1% with LATUDA 20 to 120 mg compared to 0.9% with placebo.
Pediatric Patients (10 to 17 years): In the 6-week, placebo-controlled bipolar depression study in pediatric patients 10 to 17 years, there were no reported adverse events of orthostatic hypotension or syncope.
Orthostatic hypotension, as assessed by vital signs, occurred with a frequency of 1.1% with LATUDA 20 to 80 mg/day, compared to 0.6% with placebo.
Falls: LATUDA may cause somnolence, postural hypotension, motor and sensory instability, which may lead to falls and, consequently, fractures or other injuries. For patients with diseases, conditions, or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic therapy.
Seizures: As with other antipsychotic drugs, LATUDA should be used cautiously in patients with a history of seizures or with conditions that lower the seizure threshold, e.g., Alzheimer's dementia. Conditions that lower the seizure threshold may be more prevalent in patients 65 years or older.
Schizophrenia: In adult short-term, placebo-controlled schizophrenia studies, seizures/convulsions occurred in 0.1% (2/1508) of patients treated with LATUDA compared to 0.1% (1/708) placebo-treated patients.
Bipolar Depression: Monotherapy: In the adult and pediatric 6-week, flexible-dose, placebo-controlled monotherapy bipolar depression studies, no patients experienced seizures/convulsions.
Adjunctive Therapy with Lithium or Valproate: In the adult short-term, flexible-dose, placebo-controlled adjunctive therapy bipolar depression studies, no patient experienced seizures/convulsions.
Potential for Cognitive and Motor Impairment: LATUDA, like other antipsychotics, has the potential to impair judgment, thinking or motor skills. Caution patients about operating hazardous machinery, including motor vehicles, until they are reasonably certain that therapy with LATUDA does not affect them adversely.
In clinical studies with LATUDA, somnolence included: hypersomnia, hypersomnolence, sedation and somnolence.
Schizophrenia: Adults: In short-term, placebo-controlled schizophrenia studies, somnolence was reported by 17.0% (256/1508) of patients treated with LATUDA (15.5% LATUDA 20 mg, 15.6% LATUDA 40 mg, 15.2% LATUDA 80 mg, 26.5% LATUDA 120 mg and 8.3% LATUDA 160 mg/day) compared to 7.1% (50/708) of placebo patients.
Adolescents: In the short-term, placebo-controlled adolescent schizophrenia study, somnolence was reported by 14.5% (31/214) of patients treated with LATUDA (15.5% LATUDA 40 mg and 13.5% LATUDA 80 mg,/day) compared to 7.1% (8/112) of placebo patients.
Bipolar Depression: Adults: Monotherapy: In the adult short-term, flexible-dosed, placebo-controlled monotherapy bipolar depression study, somnolence was reported by 7.3% (12/164) and 13.8% (23/167) with LATUDA 20 to 60 mg and 80 to120 mg, respectively compared to 6.5% (11/168) of placebo patients.
Adjunctive Therapy with Lithium or Valproate: In the adult short-term, flexible-dosed, placebo-controlled adjunctive therapy bipolar depression studies, somnolence was reported by 11.4% (41/360) of patients treated with LATUDA 20-120 mg compared to 5.1% (17/334) of placebo patients.
Pediatric Patients (10 to 17 years): In the 6-week, placebo-controlled bipolar depression study in pediatric patients 10 to 17 years, somnolence was reported by 11.4% (20/175) of patients treated with LATUDA 20 to 80 mg/day compared to 5.8% (10/172) of placebo treated patients.
Body Temperature Dysregulation: Disruption of the body's ability to reduce core body temperature has been attributed to antipsychotic agents. Appropriate care is advised when prescribing LATUDA for patients who will be experiencing conditions that may contribute to an elevation in core body temperature, e.g., exercising strenuously, exposure to extreme heat, receiving concomitant medication with anticholinergic activity, or being subject to dehydration.
Activation of Mania/Hypomania: Antidepressant treatment can increase the risk of developing a manic or hypomanic episode, particularly in patients with bipolar disorder. Monitor patients for the emergence of such episodes.
In the adult bipolar depression monotherapy and adjunctive therapy (with lithium or valproate) studies, less than 1% of subjects in the LATUDA and placebo groups developed manic or hypomanic episodes.
Dysphagia: Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Aspiration pneumonia is a common cause of morbidity and mortality in elderly patients, in particular those with advanced Alzheimer's dementia. LATUDA and other antipsychotic drugs should be used cautiously in patients at risk for aspiration pneumonia.
Neurological Adverse Reactions in Patients with Parkinson's Disease or Dementia with Lewy Bodies: Patients with Parkinson's Disease or Dementia with Lewy Bodies are reported to have an increased sensitivity to antipsychotic medication. Manifestations of this increased sensitivity include confusion, obtundation, postural instability with frequent falls, extrapyramidal symptoms, and clinical features consistent with the neuroleptic malignant syndrome.
Drug Abuse and Dependence: LATUDA is not a controlled substance. LATUDA has not been systematically studied in humans for its potential for abuse or physical dependence or its ability to induce tolerance. While clinical studies with LATUDA did not reveal any tendency for drug-seeking behavior, these observations were not systematic and it is not possible to predict the extent to which a CNS-active drug will be misused, diverted and/or abused once it is marketed. Patients should be evaluated carefully for a history of drug abuse, and such patients should be observed carefully for signs of LATUDA misuse or abuse (e.g., development of tolerance, drug-seeking behavior, increases in dose).
Renal Impairment: Reduce the maximum recommended dosage in patients with moderate or severe renal impairment (CLcr<50 mL/minute). Patients with impaired renal function (CLcr<50 mL/minute) had higher exposure to lurasidone than patients with normal renal function [see Pharmacology under Actions]. Greater exposure may increase the risk of LATUDA-associated adverse reactions [see Dosage & Administration]
Hepatic Impairment: Reduce the maximum recommended dosage in patients with moderate to severe hepatic impairment (Child-Pugh score ≥7). Patients with moderate to severe hepatic impairment (Child-Pugh score ≥7) generally had higher exposure to lurasidone than patients with normal hepatic function [see Pharmacology under Actions]. Greater exposure may increase the risk of LATUDA-associated adverse reactions [see Dosage & Administration].
Other Specific Populations: No dosage adjustment for LATUDA is required on the basis of a patient's sex, race, or smoking status [see Pharmacology: Pharmacokinetics under Actions].
Use in Children: Schizophrenia: The safety and effectiveness of LATUDA 40-mg/day and 80-mg/day for the treatment of schizophrenia in adolescents (13 to 17 years) was established in a 6-week, placebo-controlled clinical study in 326 adolescent patients [see Dosage & Administration, Adverse Reactions].
The safety and effectiveness of LATUDA has not been established in pediatric patients less than 13 years of age with schizophrenia.
Bipolar Depression: The safety and effectiveness of LATUDA 20 to 80 mg/day for the treatment of bipolar depression in pediatric patients (10 to 17 years) was evaluated in a 6-week, placebo-controlled clinical study in 347 pediatric patients [see Dosage & Administration, Adverse Reactions].
The safety and effectiveness of LATUDA has not been established in pediatric patients less than 13 years of age with bipolar depression.
Irritability Associated with Autistic Disorder: The effectiveness of LATUDA in pediatric patients for the treatment of irritability associated with autistic disorder has not been established.
Efficacy was not demonstrated in a 6-week study evaluating LATUDA 20 mg/day and 60 mg/day for the treatment of pediatric patients 6 to 17 years of age with irritability associated with autistic disorder diagnosed by Diagnostic and Statistical Manual of Mental Disorders, 4th Ed., Text Revision [DSM-IV-TR] criteria. The primary objective of the study as measured by improvement from Baseline in the irritability subscale of the Aberrant Behavior Checklist (ABC) at Endpoint (Week 6) was not met. A total of 149 patients were randomized to LATUDA or placebo. Vomiting occurred at a higher rate than reported in other LATUDA studies (4/49 or 8% for 20mg, 14/51 or 27% for 60mg, and 2/49 or 4% for placebo), particularly in children ages 6 to 12 (13 out of 18 patients on LATUDA with vomiting).
In a long-term, open-label study that enrolled pediatric patients (age 6 to 17 years) with schizophrenia, bipolar depression, or autistic disorder from three short-term, placebo-controlled trials, 54% (378/701) received lurasidone for 104 weeks. There was one adverse event in this trial that was considered possibly drug-related and has not been reported in adults receiving lurasidone: a 10 years old male experienced a prolonged, painful erection, consistent with priapism, that led to treatment discontinuation.
In this trial, the mean increase in height from open-label baseline to Week 104 was 4.94 cm. To adjust for normal growth, z-scores were derived (measured in standard deviations [SD]), which normalize for the natural growth of children and adolescents by comparisons to age- and sex-matched population standards. A z-score change <0.5 SD is considered not clinically significant. In this trial, the mean change in height z-score from open-label baseline to Week 104 was +0.05 SD, indicating minimal deviation from the normal growth curve.
Juvenile animal studies: Adverse effects were seen on growth, physical and neurobehavioral development at doses as low as 0.2 times the MRHD based on mg/m
2. Lurasidone was orally administered to rats from postnatal days 21 through 91 (this period corresponds to childhood, adolescence, and young adulthood in humans) at doses of 3, 30, and 150 (males) or 300 (females) mg/kg/day which are 0.2 to 10 times (males) and 20 times (females) the maximum recommended adult human dose (MRHD) of 160 mg/day based on mg/m
2. The adverse effects included dose-dependent decreases in femoral length, bone mineral content, body and brain weights at 2 times the MRHD in both sexes, and motor hyperactivity at 0.2 and 2 times the MRHD in both sexes based on mg/m
2. In females, there was a delay in attainment of sexual maturity at 2 times the MRHD, associated with decreased serum estradiol. Mortality occurred in both sexes during early post-weaning period and some of the male weanlings died after only 4 treatments at doses as low as 2 times the MRHD based on mg/m
2. Histopathological findings included increased colloid in the thyroids and inflammation of the prostate in males at 10 times MRHD based on mg/m
2 and mammary gland hyperplasia, increased vaginal mucification, and increased ovarian atretic follicles at doses as low as 0.2 times the MRHD based on mg/m
2. Some of these findings were attributed to transiently elevated serum prolactin which was seen in both sexes at all doses. However, there were no changes at any dose level in reproductive parameters (fertility, conception indices, spermatogenesis, estrous cycle, gestation length, parturition, number of pups born). The no effect dose for neurobehavioral changes in males is 0.2 times the MRHD based on mg/m
2 and could not be determined in females. The no effect dose for growth and physical development in both sexes is 0.2 times the MRHD based on mg/m
2.
Use in the Elderly: Clinical studies with LATUDA did not include sufficient numbers of patients aged 65 and older to determine whether or not they respond differently from younger patients. In elderly patients with psychosis (65 to 85), LATUDA concentrations (20 mg/day) were similar to those in young subjects. It is unknown whether dose adjustment is necessary on the basis of age alone.
Elderly patients with dementia-related psychosis treated with LATUDA are at an increased risk of death compared to placebo. LATUDA is not approved for the treatment of patients with dementia-related psychosis [see Precautions].



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image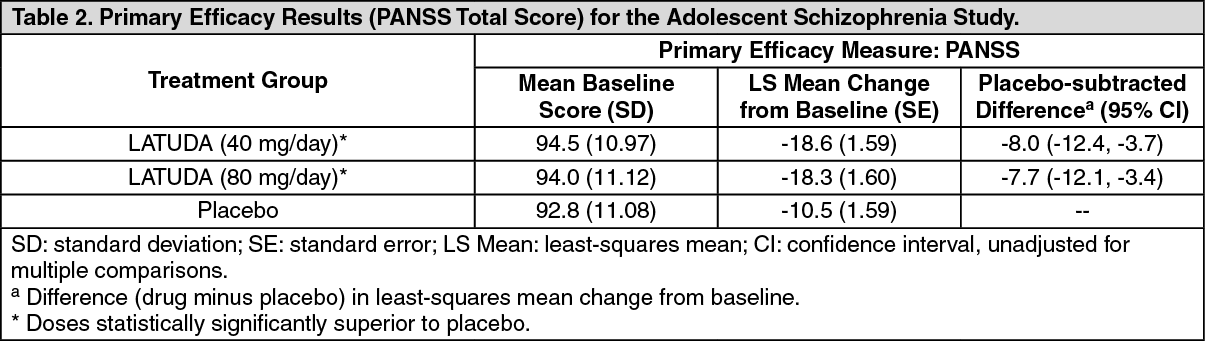 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image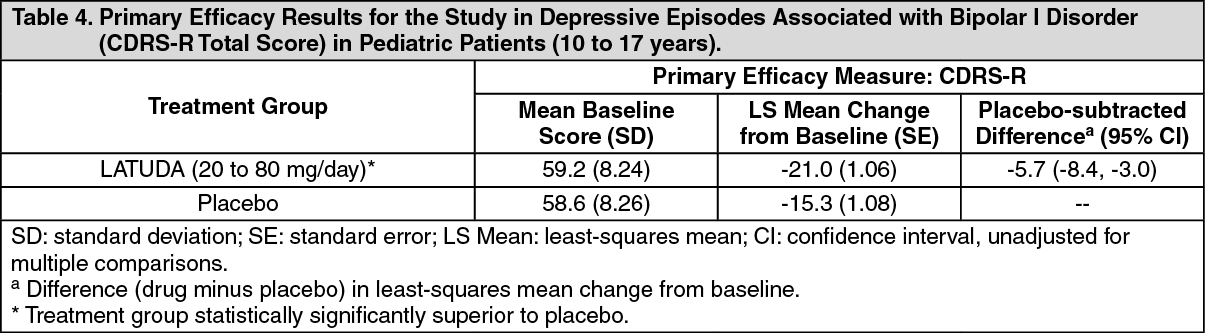 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image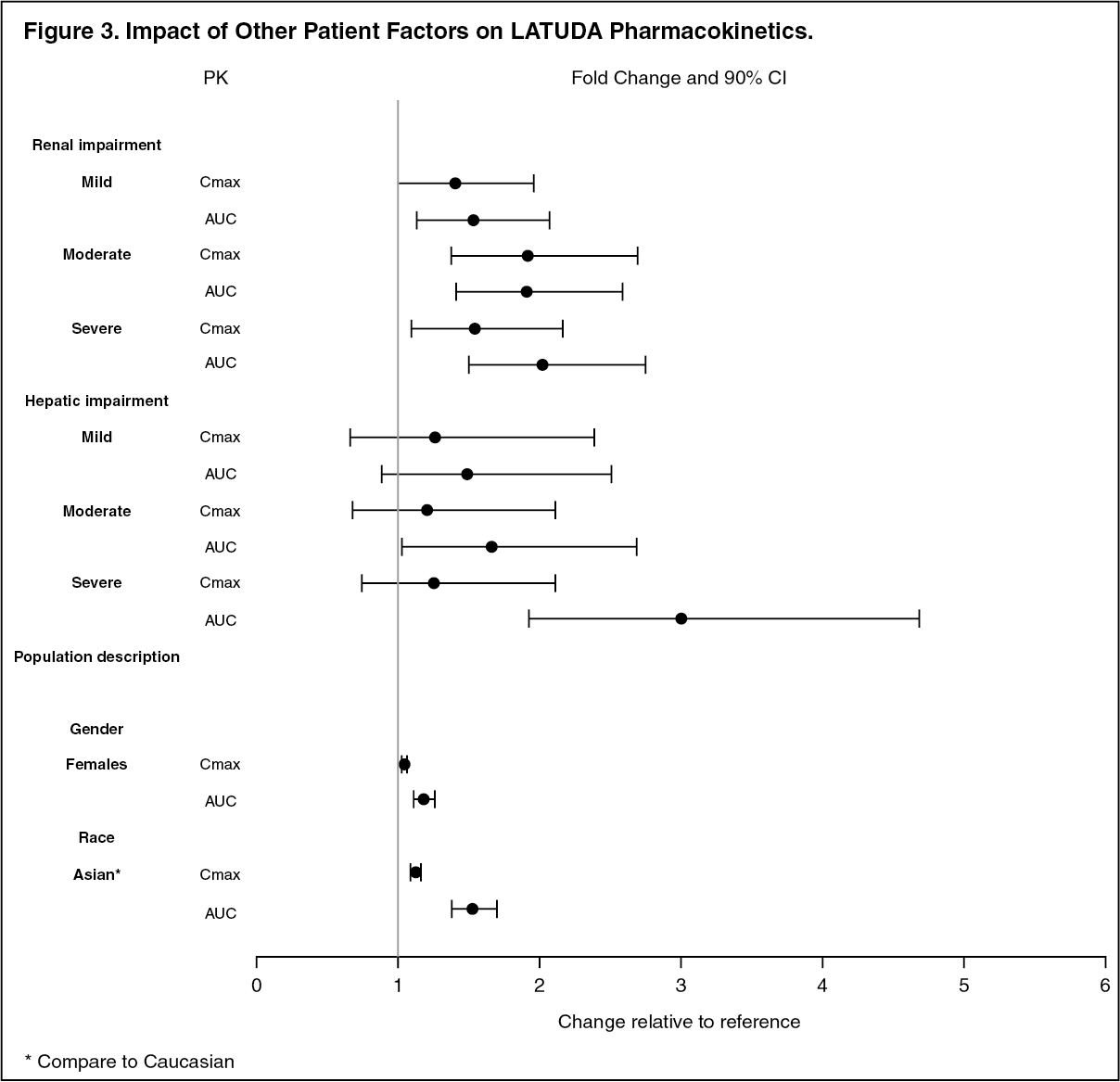 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image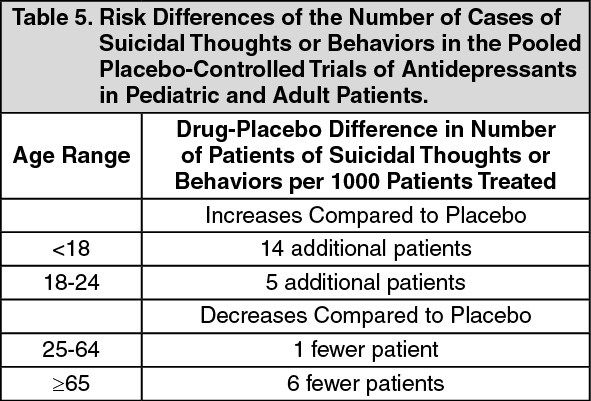 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image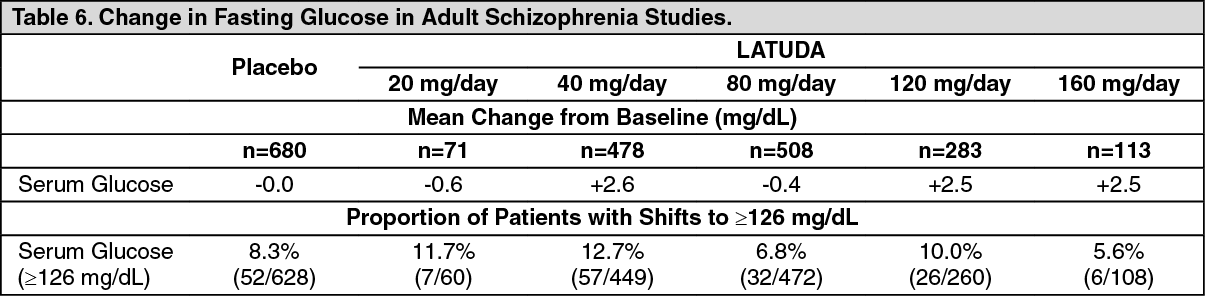 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image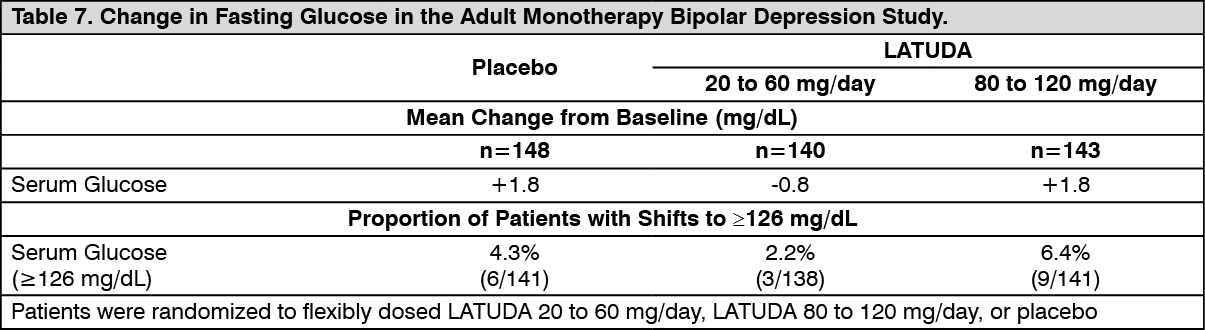 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image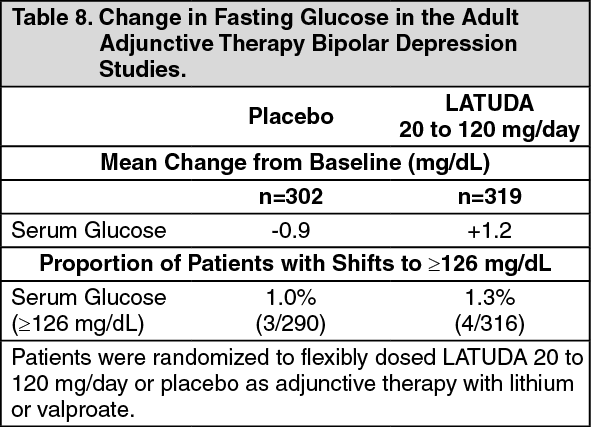 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image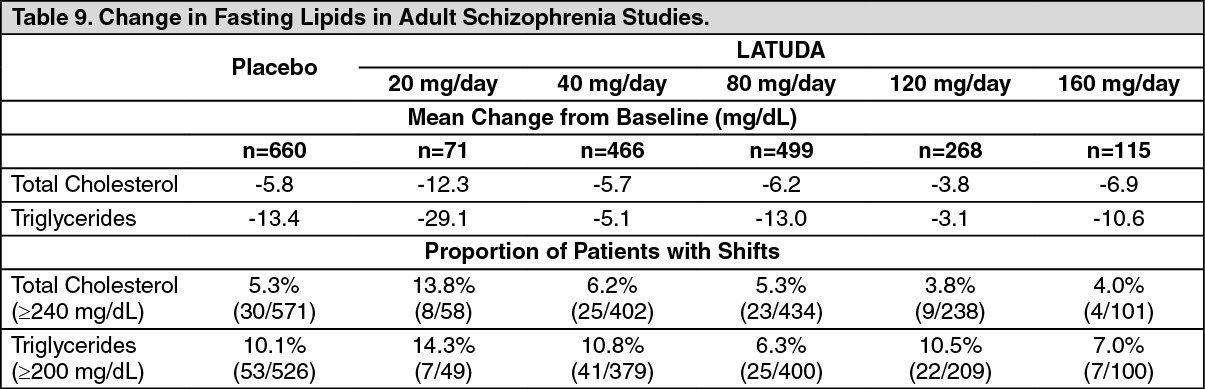 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image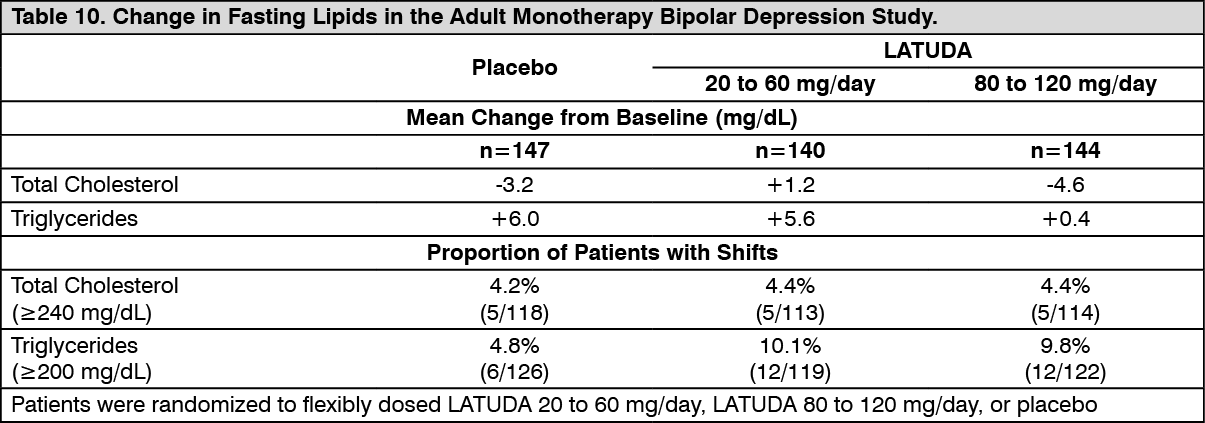 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image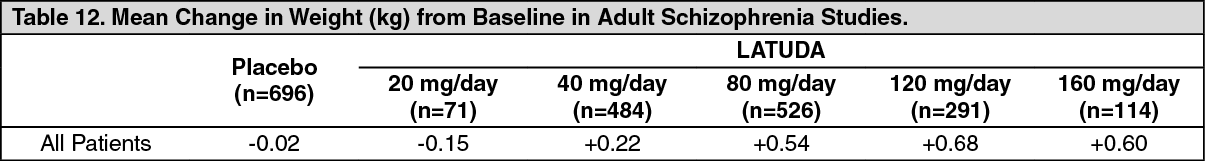 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image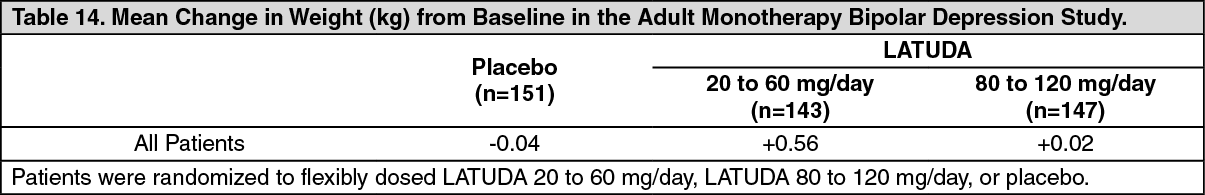 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image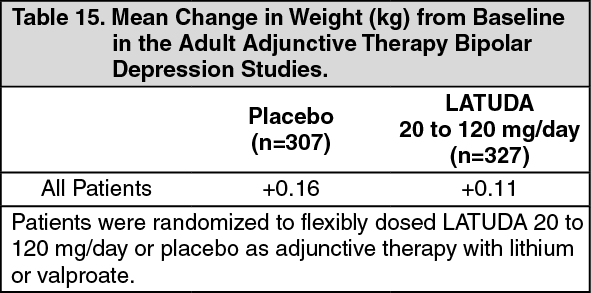 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image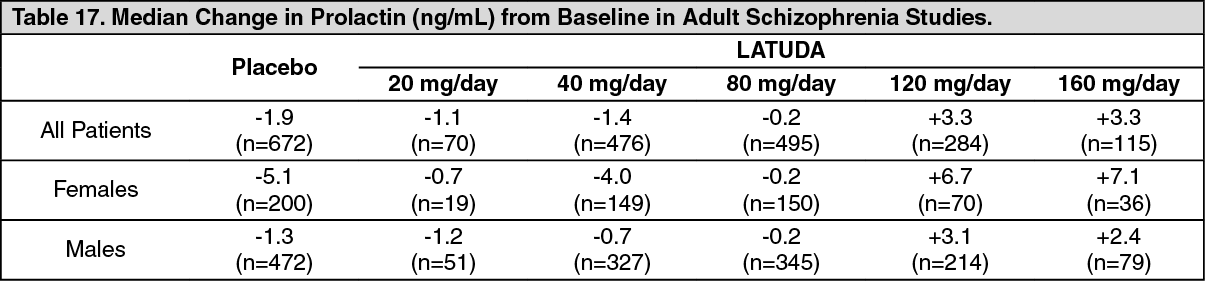 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image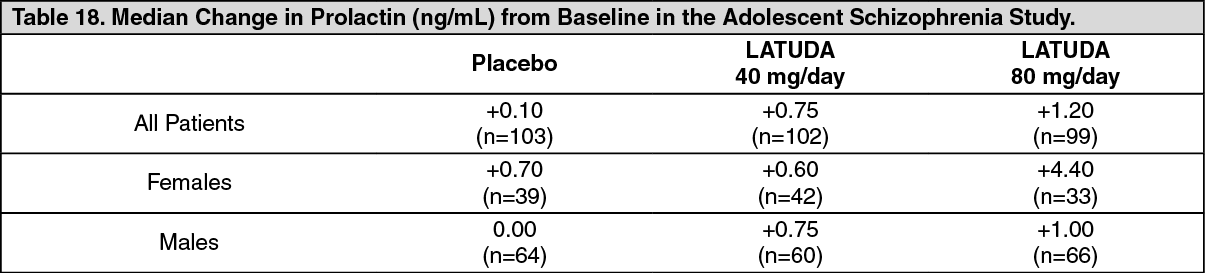 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image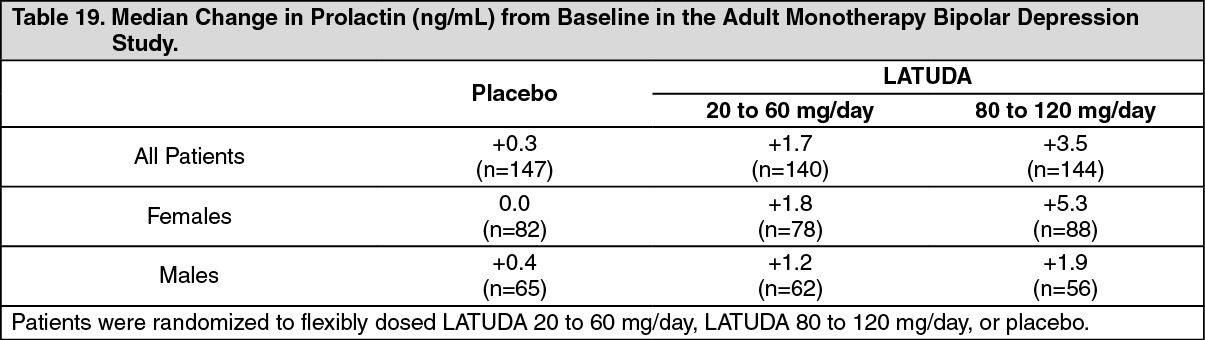 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image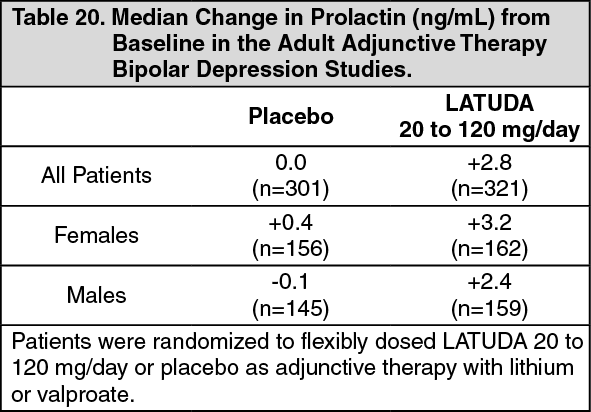 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image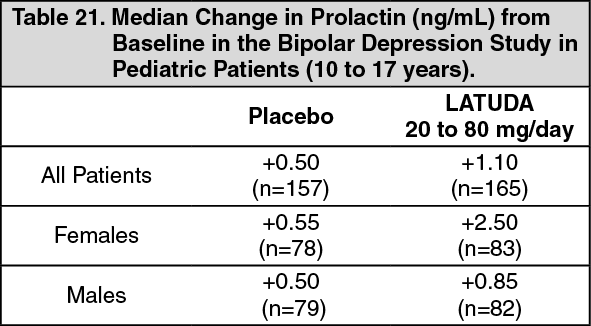 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image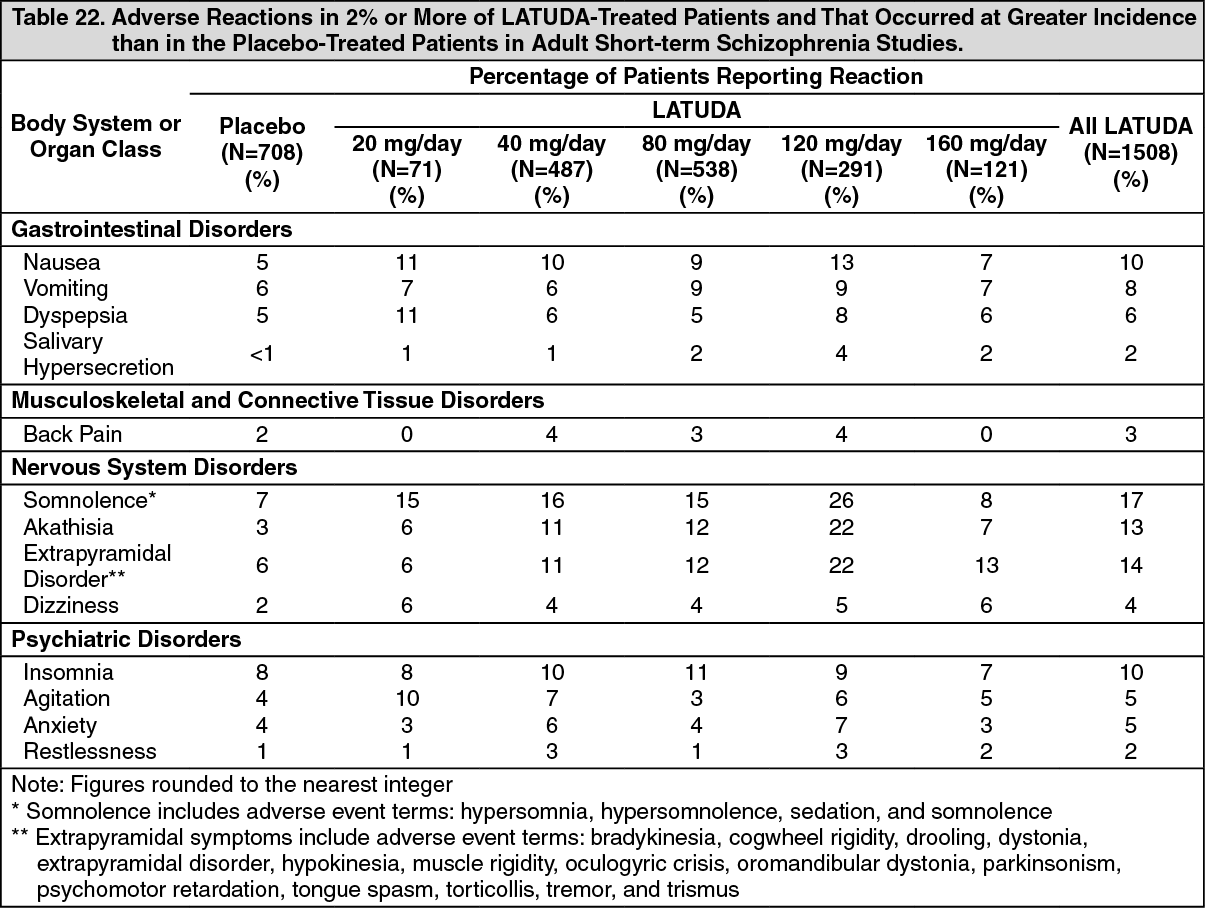 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image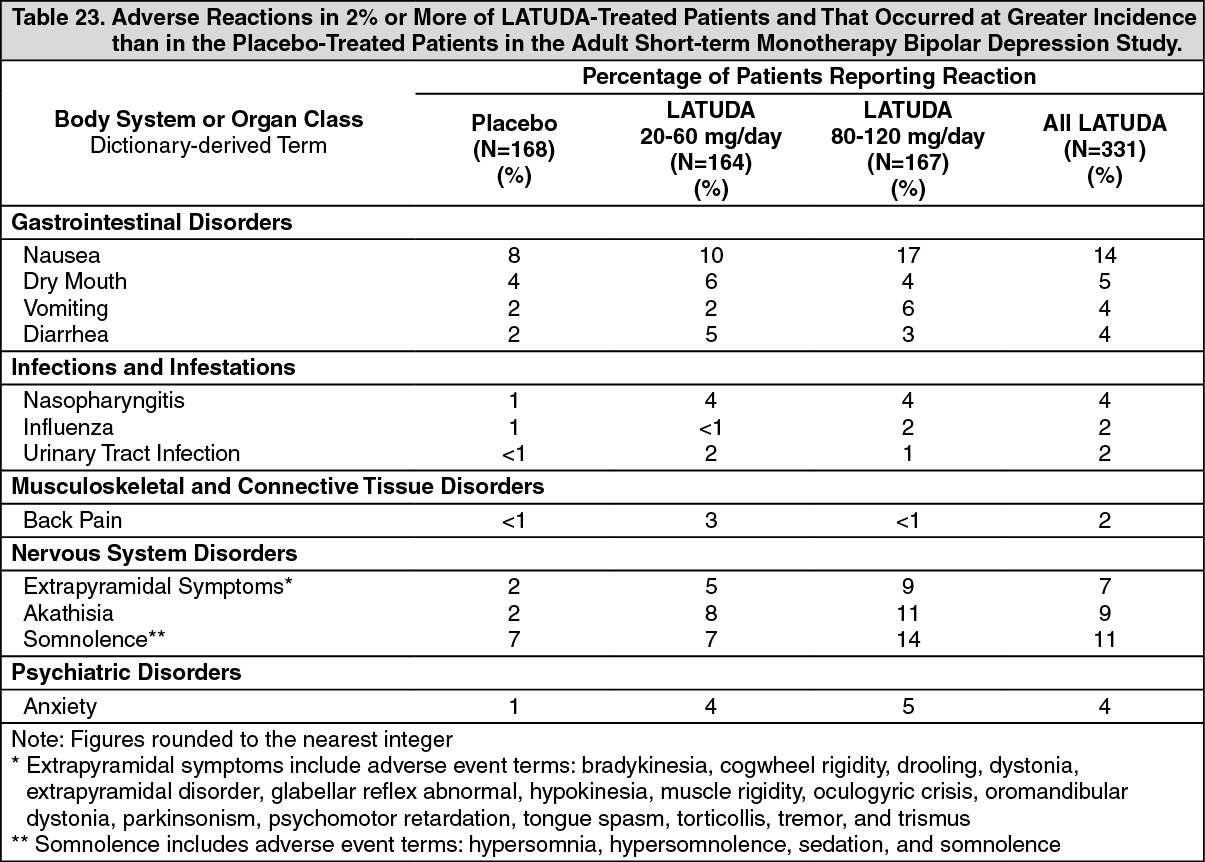 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image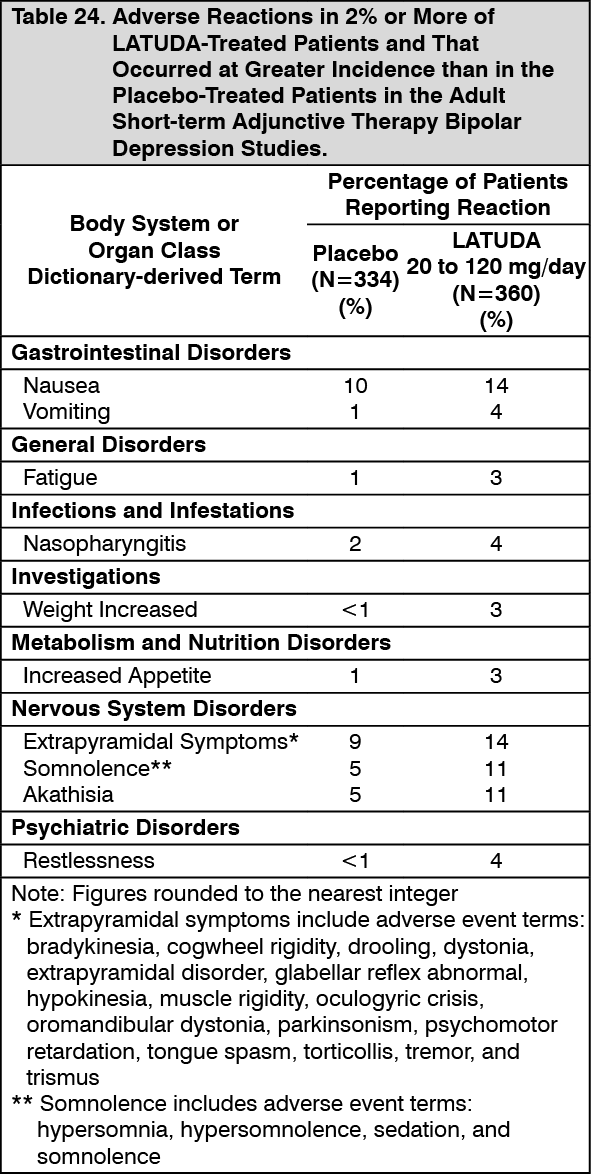 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image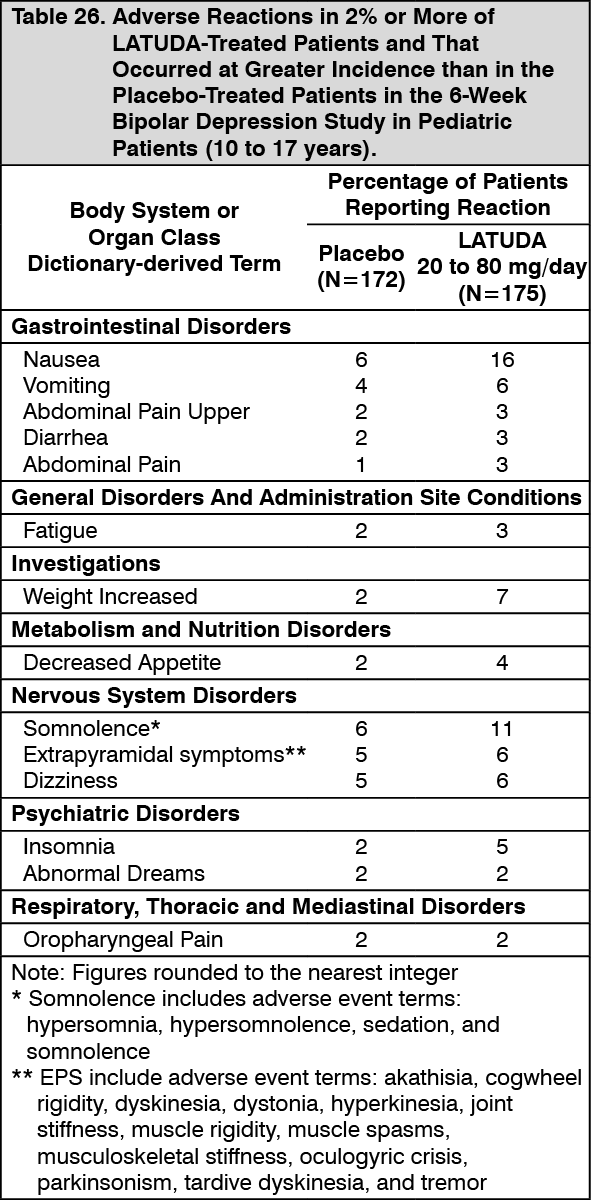 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image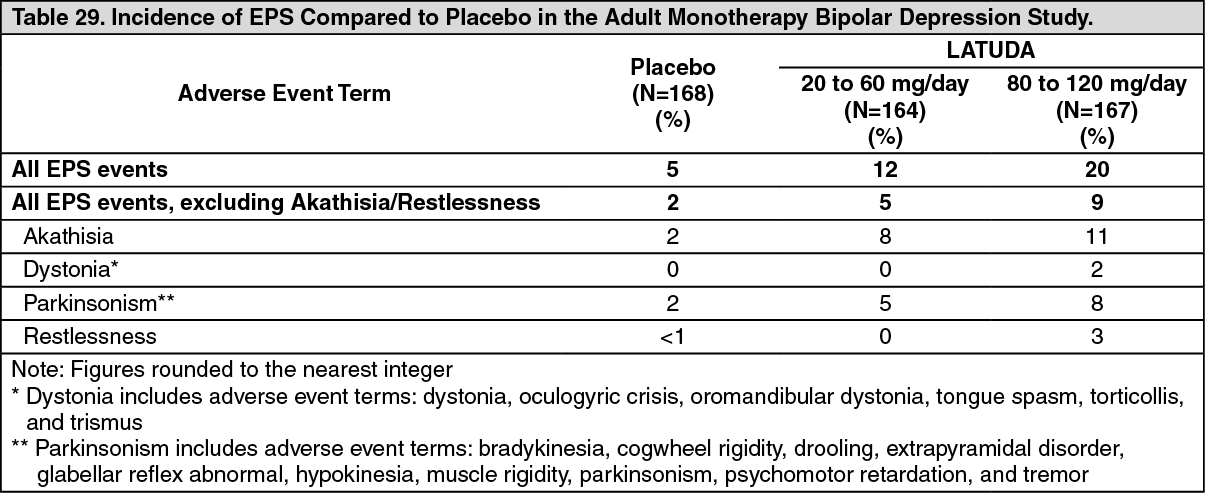 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image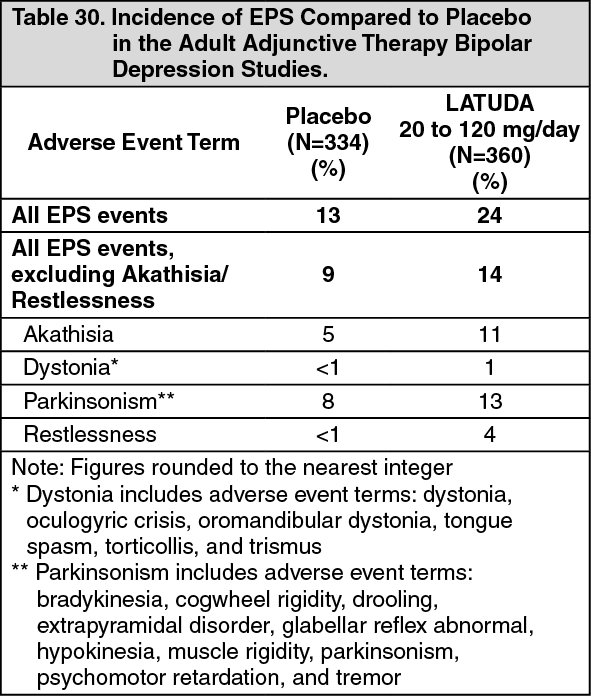 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image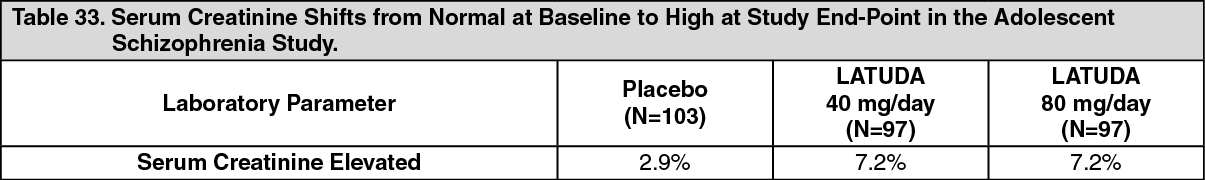 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image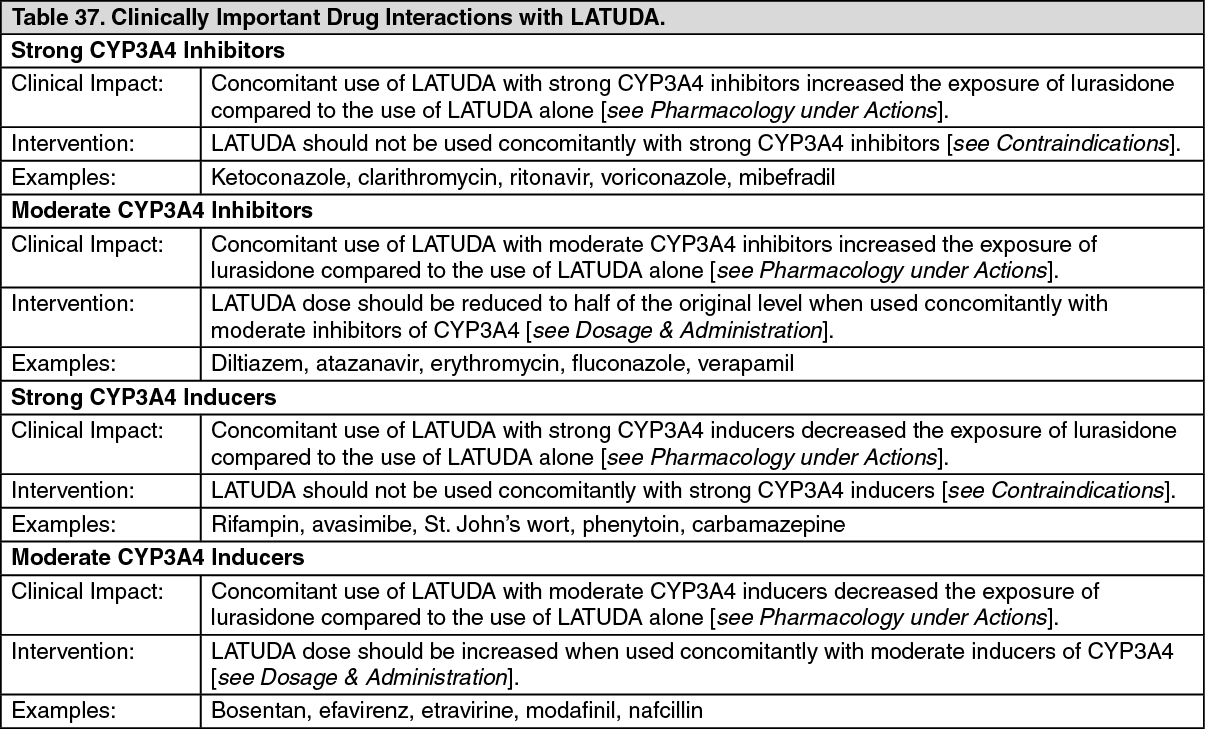 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
