Pharmacology: Mechanism of Action: Expression of programmed cell death ligand-1 (PD-L1) can be induced by inflammatory signals (e.g., IFN-gamma) and can be expressed on both tumor cells and tumor-associated immune cells in the tumor microenvironment. PD-L1 blocks T-cell function and activation through interaction with PD-1 and CD80 (B7.1). By binding to its receptors, PD-L1 reduces cytotoxic T-cell activity, proliferation, and cytokine production.
Durvalumab is a human immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that binds to PD-L1 and blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1). Blockade of PD-L1/PD-1 and PD-L1/CD80 interactions releases the inhibition of immune responses, without inducing antibody dependent cell-mediated cytotoxicity (ADCC).
PD-L1 blockade with durvalumab led to increased T-cell activation in vitro and decreased tumor size in co-engrafted human tumor and immune cell xenograft mouse models.
Pharmacodynamics: The steady state AUC, C
trough, and C
max in patients administered with 1500 mg every 4 weeks are 6% higher, 19% lower, and 55% higher than those administered with 10 mg/kg every 2 weeks, respectively. Based on the modeling of pharmacokinetic data and exposure relationships for safety, there are no anticipated clinically meaningful differences in efficacy and safety for the doses of 1500 mg every 4 weeks compared to 10 mg/kg every 2 weeks in patients weighing > 30 kg with NSCLC.
Clinical Studies: Non-Small Cell Lung Cancer (NSCLC): The efficacy of IMFINZI was evaluated in the PACIFIC study (NCT02125461), a multicenter, randomized, double-blind, placebo-controlled study in patients with unresectable Stage III NSCLC who completed at least 2 cycles of concurrent platinum-based chemotherapy and definitive radiation within 42 days prior to initiation of the study drug and had a WHO performance status of 0 or 1. The study excluded patients who had progressed following concurrent chemoradiation, patients with active or prior documented autoimmune disease within 2 years of initiation of the study or patients with medical conditions that required systemic immunosuppression. Randomization was stratified by sex, age (< 65 years vs. ≥ 65 years) and smoking history (smoker vs. non-smoker). Patients were randomized 2:1 to receive IMFINZI 10 mg/kg or placebo intravenously every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed RECIST 1.1-defined progression. Assessment of tumor status was performed every 8 weeks. The major efficacy outcome measures were progression-free survival (PFS) as assessed by a BICR RECIST 1.1 and overall survival (OS). Additional efficacy outcome measures included ORR and DoR assessed by BICR.
A total of 713 patients were randomized: 476 patients to the IMFINZI arm and 237 to the placebo arm. The study population characteristics were: median age of 64 years (range: 23 to 90); 70% male; 69% White and 27% Asian; 16% current smokers, 75% former smokers and 9% never smokers; 51% WHO performance status of 1; 53% with Stage IIIA and 45% were Stage IIIB; 46% with squamous and 54% with non-squamous histology. All patients received definitive radiotherapy as per protocol, of which 92% received a total radiation dose of 54 Gy to 66 Gy; 99% of patients received concomitant platinum-based chemotherapy (55% cisplatin-based, 42% carboplatin-based chemotherapy and 2% switched between cisplatin and carboplatin).
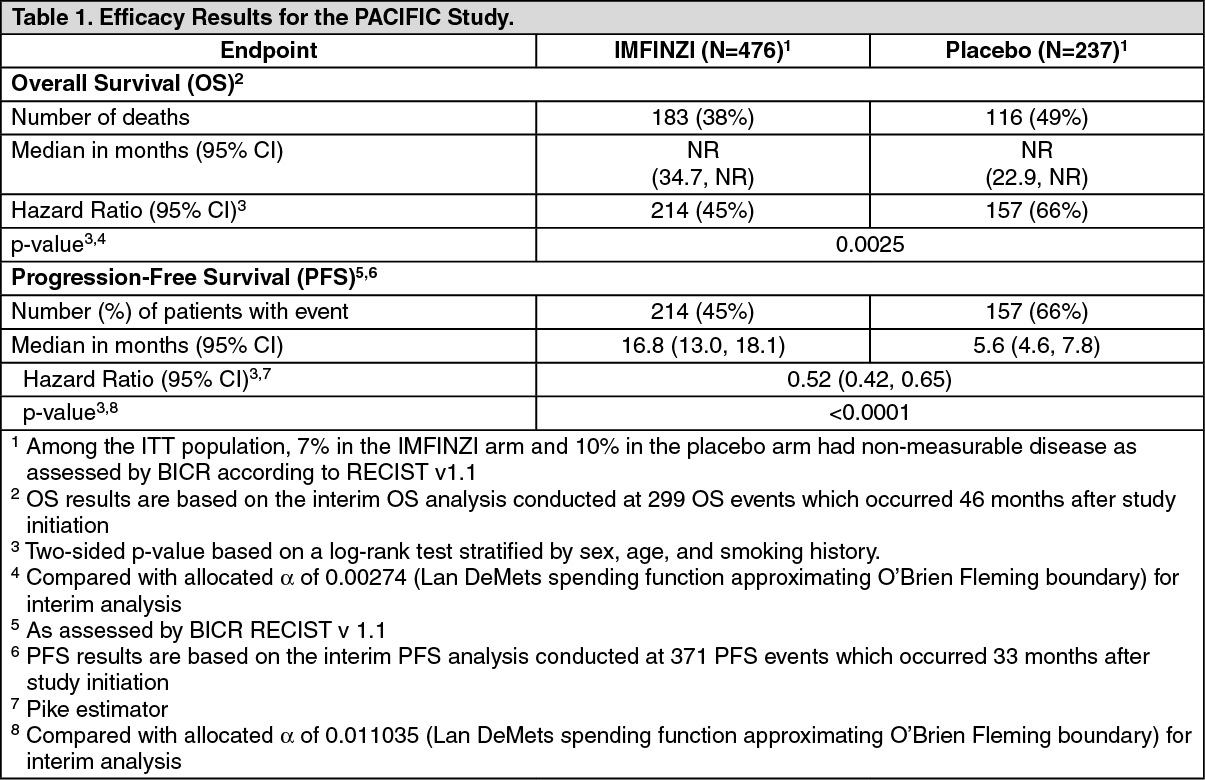
At a pre-specified interim analysis for OS based on 299 events (61% of total planned events), the study demonstrated a statistically significant improvement in OS in patients randomized to IMFINZI compared to placebo. The pre-specified interim PFS analysis based on 371 events (81% of total planned events) demonstrated a statistically significant improvement in PFS in patients randomized to IMFINZI compared to placebo. Table 1 and Figure 1 summarizes the efficacy results for PACIFIC. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Small Cell Lung Cancer (SCLC): The efficacy of IMFINZI in combination with etoposide and either carboplatin or cisplatin in previously untreated ES-SCLC was investigated in CASPIAN, a randomized, multicenter, active-controlled, open-label, trial (NCT03043872). Eligible patients had WHO Performance Status of 0 or 1 and were suitable to receive a platinum-based chemotherapy regimen as first-line treatment for SCLC. Patients with asymptomatic or treated brain metastases were eligible. Choice of platinum agent was at the investigator's discretion, taking into consideration the calculated creatinine clearance. Patients with history of chest radiation therapy; a history of active primary immunodeficiency; autoimmune disorders including paraneoplastic syndrome; active or prior documented autoimmune or inflammatory disorders; use of systemic immunosuppressants within 14 days before the first dose of the treatment except physiological dose of systemic corticosteroids were ineligible.
Randomization was stratified by the planned platinum-based therapy in cycle 1 (carboplatin or cisplatin).
The evaluation of efficacy for ES-SCLC relied on Arms 2 and 3: IMFINZI 1500 mg, and investigator's choice of carboplatin (AUC 5 or 6 mg/mL/min) or cisplatin (75-80 mg/m
2) on Day 1 and etoposide (80-100 mg/m
2) intravenously on Days 1, 2, and 3 of each 21-day cycle for 4 cycles, followed by IMFINZI 1500 mg every 4 weeks until disease progression or unacceptable toxicity, or; Investigator's choice of carboplatin (AUC 5 or 6 mg/mL/min) or cisplatin (75-80 mg/m
2) on Day 1 and etoposide (80-100 mg/m
2) intravenously on Days 1, 2, and 3 of each 21-day cycle, up to 6 cycles. After completion of chemotherapy, prophylactic cranial irradiation (PCI) as administered per investigator discretion.
Administration of IMFINZI as a single agent was permitted beyond disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator. The major efficacy outcome measure was overall survival (OS) of IMFINZI plus chemotherapy vs. chemotherapy alone. Additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR), per RECIST v1.1.
The study population characteristics were: median age of 63 years (range: 28 to 82); 40% age 65 or older; 70% male; 84% White, 15% Asian, and 0.9% Black; 65% WHO/ECOG PS of 1; and 93% were former/current smokers. Ninety percent of patients had Stage IV disease and 10% had brain metastasis at baseline. A total of 25% of the patients received cisplatin and 74% of the patients received carboplatin. In the chemotherapy alone arm, 57% of the patients received 6 cycles of chemotherapy, and 8% of the patients received PCI.
The OS results for are summarized in Table 2 and Figure 2. (See Table 2 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Investigator-assessed PFS (96% of total planned events) showed a HR of 0.78 (95% CI: 0.65, 0.94), with median PFS of 5.1 months (95% CI: 4.7, 6.2) in the IMFINZI plus chemotherapy arm and 5.4 months (95% CI: 4.8, 6.2) in the chemotherapy alone arm. The investigator-assessed confirmed ORR was 68% (95% CI: 62%, 73%) in the IMFINZI plus chemotherapy arm and 58% (95% CI: 52%, 63%) in the chemotherapy alone arm.
In the exploratory subgroup analyses of OS based on the planned platinum chemotherapy received at cycle 1, the HR was 0.70 (95% CI 0.55, 0.89) in patients who received carboplatin, and the HR was 0.88 (95% CI 0.55, 1.41) in patients who received cisplatin.
Biliary Tract Cancer (BTC): The efficacy of IMFINZI in combination with gemcitabine and cisplatin in patients with locally advanced or metastatic BTC was investigated in TOPAZ-1 (NCT03875235), a randomized, double-blind, placebo-controlled, multicenter trial that enrolled 685 patients with histologically confirmed locally advanced unresectable or metastatic BTC who have not previously received systemic therapy. Patients with recurrent disease > 6 months after surgery and/or completion of adjuvant therapy were eligible. Patients had an ECOG Performance status of 0 and 1 and least one target lesion by RECIST 1.1. Patients with ampullary carcinoma; active or prior documented autoimmune or inflammatory disorders; HIV infection or active infections, including tuberculosis or hepatitis C; current or prior use of immunosuppressive medication within 14 days before the first dose of IMFINZI were ineligible.
Randomization was stratified by disease status (recurrent vs. initially unresectable) and primary tumor location (intrahepatic cholangiocarcinoma [ICCA] vs. extrahepatic cholangiocarcinoma [ECCA] vs. gallbladder cancer [GBC]). Patients were randomized 1:1 to receive: IMFINZI 1500 mg on Day 1+ gemcitabine 1000 mg/m2 and cisplatin 25 mg/m2 on Days 1 and 8 of each 21-day cycle up to 8 cycles, followed by IMFINZI 1500 mg every 4 weeks, or; Placebo on Day 1+ gemcitabine 1000 mg/m2 and cisplatin 25 mg/m2 on Days 1 and 8 of each 21-day cycle up to 8 cycles, followed by placebo every 4 weeks.
Treatment with IMFINZI or placebo continued until disease progression, or unacceptable toxicity. Treatment beyond disease progression was permitted if the patient was clinically stable and deriving clinical benefit as determined by the investigator.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were investigator-assessed progression-free survival (PFS), objective response rate (ORR) and duration of response (DoR). Tumor assessments were conducted every 6 weeks for the first 24 weeks after the date of randomization, and then every 8 weeks until confirmed objective disease progression.
The study population characteristics were: 50% male, median age of 64 years (range 20-85), 47% age 65 or older; 56% Asian, 37% White, 2% Black or African American, 0.1% American Indian or Alaskan Native, and 4% other; 51% had an ECOG PS of 1; primary tumor location was ICCA 56%, ECCA 18% and GBC 25%); 20% of patients had recurrent disease; 86% of patients had metastatic and 14% had locally advanced disease.
At a pre-specified interim analysis, the trial demonstrated a statistically significant improvement in OS and PFS in patients randomized to IMFINZI in combination with chemotherapy compared to placebo in combination with chemotherapy. Table 3 summarizes the efficacy results for TOPAZ-1. (See Table 3.)
Click on icon to see table/diagram/image
The investigator-assessed ORR was 27% (95% CI: 22%-32%) in the IMFINZI plus chemotherapy arm and 19% (95% CI: 15%-23%) in the chemotherapy alone arm. (See Figure 3.)
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics of durvalumab as a single agent was studied in patients with doses ranging from 0.1 mg/kg (0.01 times the approved recommended dosage) to 20 mg/kg (2 times the approved recommended dosage) administered once every two, three or four weeks.
PK exposure increased more than dose-proportionally at doses < 3 mg/kg (0.3 times the approved recommended dosage) and dose proportionally at doses ≥ to 3 mg/kg every 2 weeks. Steady state was achieved at approximately 16 weeks.
The pharmacokinetics of durvalumab is similar when assessed as a single agent and when in combination with chemotherapy.
Distribution: The geometric mean (% coefficient of variation [CV%]) steady state volume of distribution (V
ss) was 5.6 (18%) L.
Elimination: Durvalumab clearance decreases over time, with a mean maximal reduction (CV%) from baseline values of approximately 23% (57%) resulting in a geometric mean (CV%) steady state clearance (CL
ss) of 8.2 mL/h (39%) at day 365; the decrease in CL
ss is not considered clinically relevant. The geometric mean (CV%) terminal half-life, based on baseline CL was approximately 18 (24%) days.
Specific Populations: Age (19-96 years), body weight (31-149 kg), sex, albumin levels, lactate dehydrogenase (LDH) levels, creatinine levels, soluble PD-L1, tumor type, race, mild renal impairment (creatinine clearance (CLcr) 60 to 89 mL/min), moderate renal impairment (CLcr 30 to 59 mL/min), mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1 to 1.5x ULN and any AST), or ECOG/WHO performance status had no clinically significant effect on the pharmacokinetics of durvalumab.
The effect of severe renal impairment (CLcr 15 to 29 mL/min) or moderate hepatic impairment (bilirubin > 1.5 to 3x ULN and any AST) or severe hepatic impairment (bilirubin > 3x ULN and any AST) on the pharmacokinetics of durvalumab is unknown.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: The carcinogenic and genotoxic potential of durvalumab have not been evaluated.
Animal fertility studies have not been conducted with durvalumab. In repeat-dose toxicology studies with durvalumab in sexually mature cynomolgus monkeys of up to 3 months duration, there were no notable effects on the male and female reproductive organs.
Animal Toxicology and/or Pharmacology: In animal models, inhibition of PD-L1/PD-1 signaling increased the severity of some infections and enhanced inflammatory responses.
M. tuberculosis-infected PD-1 knockout mice exhibit markedly decreased survival compared with wild-type controls, which correlated with increased bacterial proliferation and inflammatory responses in these animals. PD-L1 and PD-1 knockout mice have also shown decreased survival following infection with lymphocytic choriomeningitis virus.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image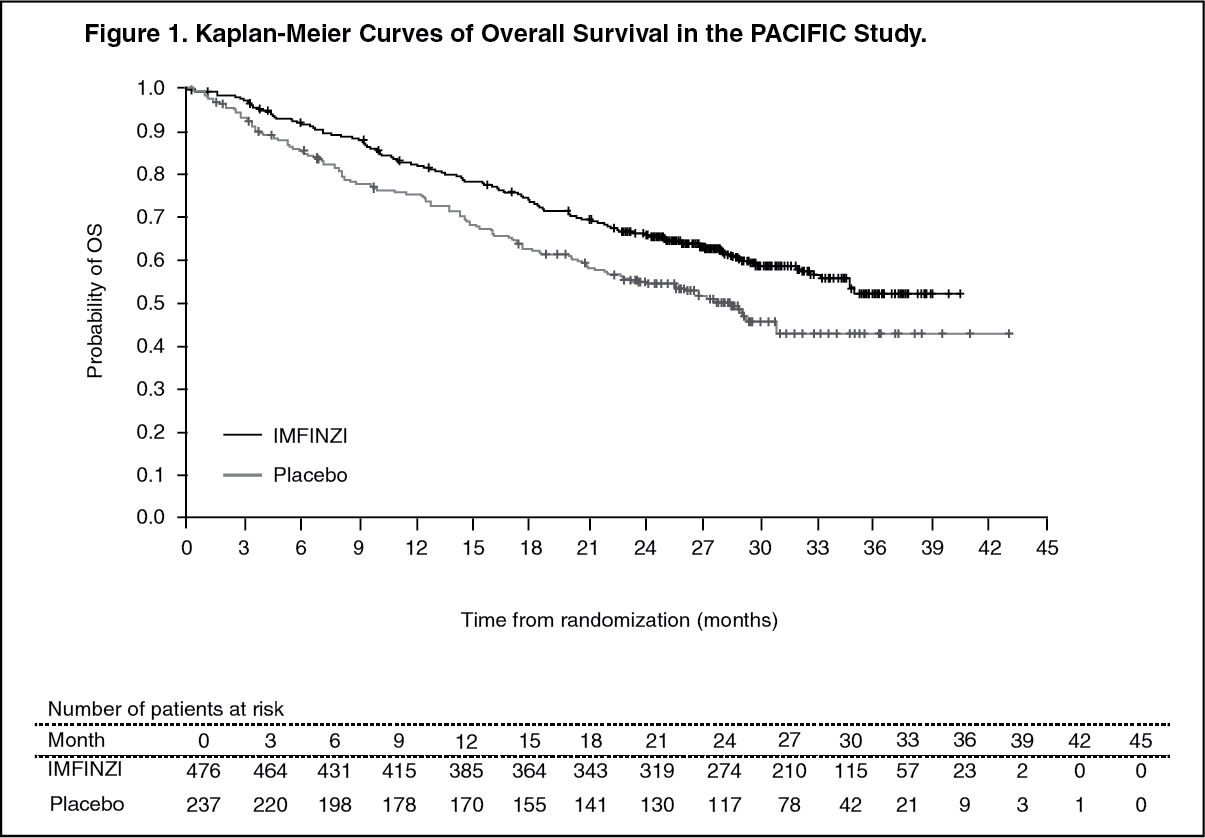 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image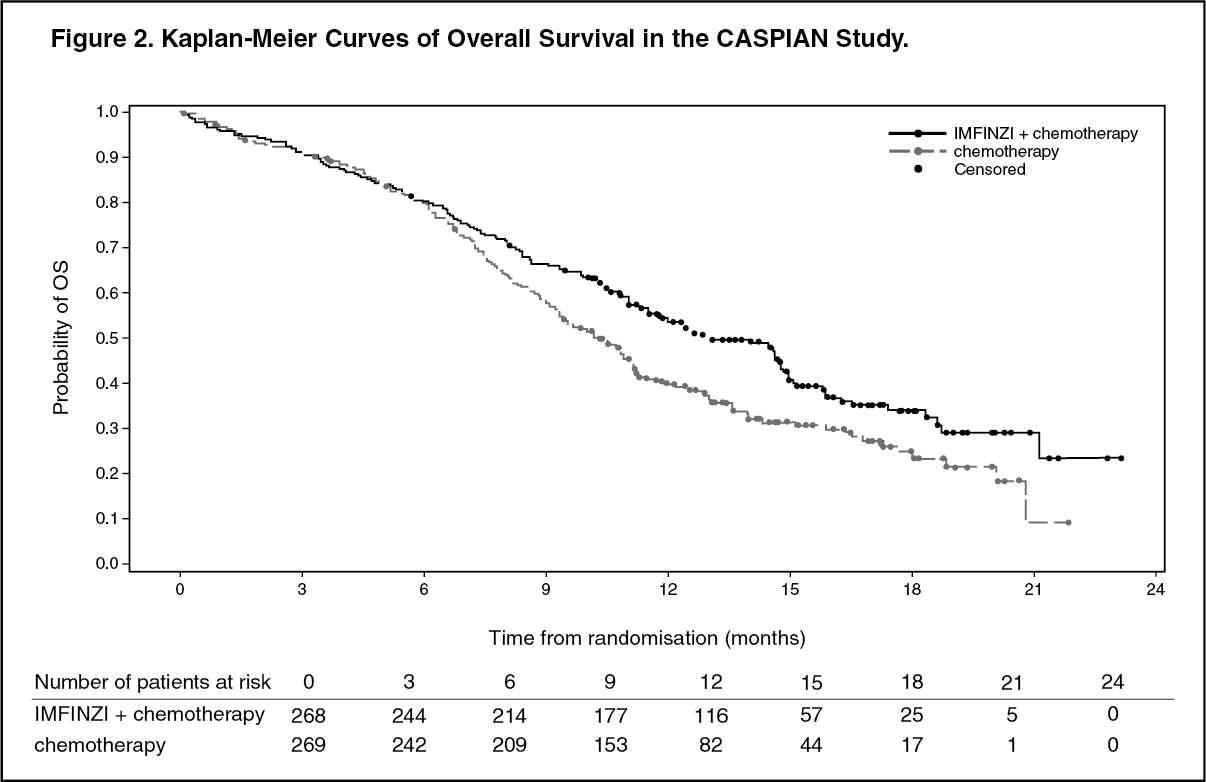 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image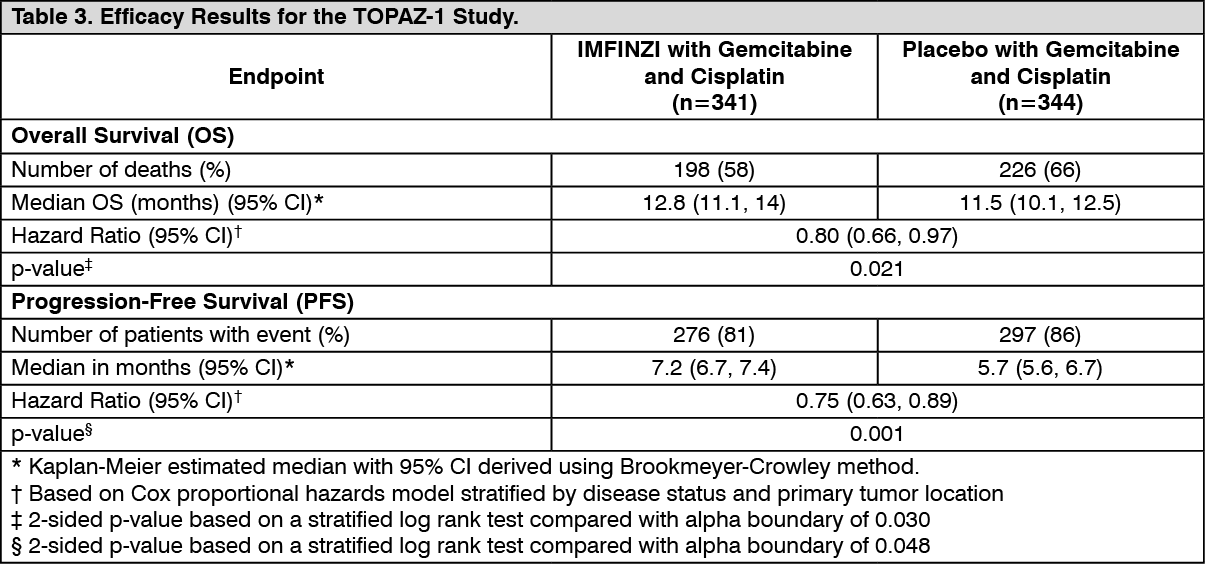 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image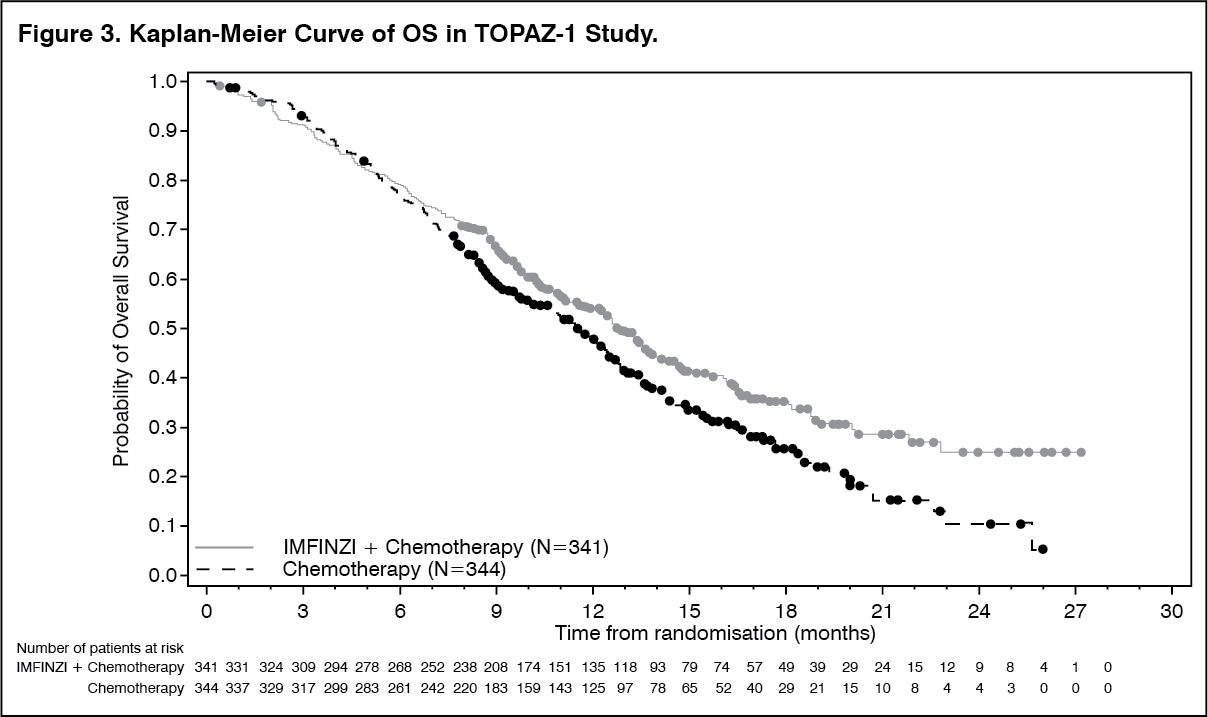 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image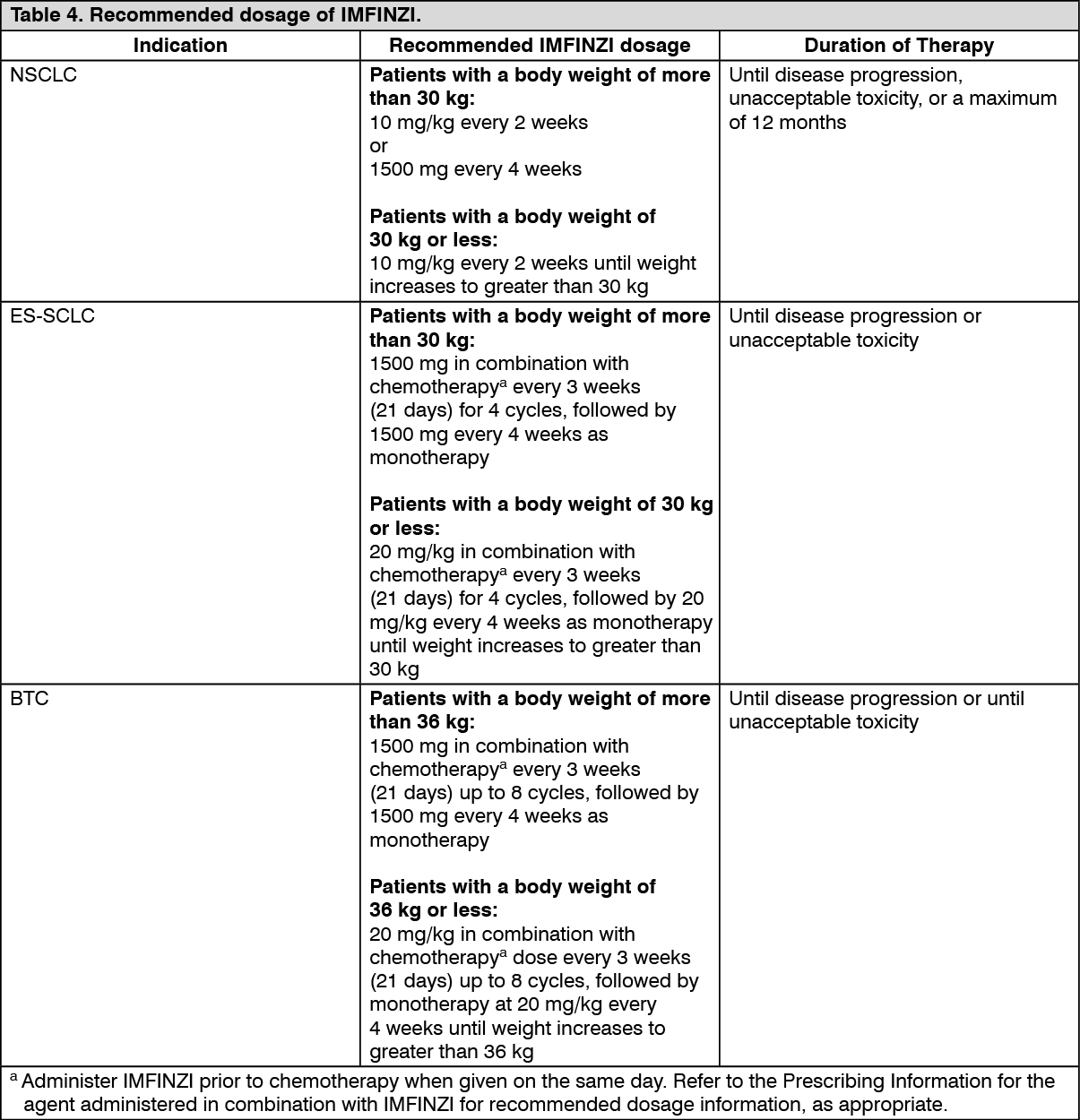 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image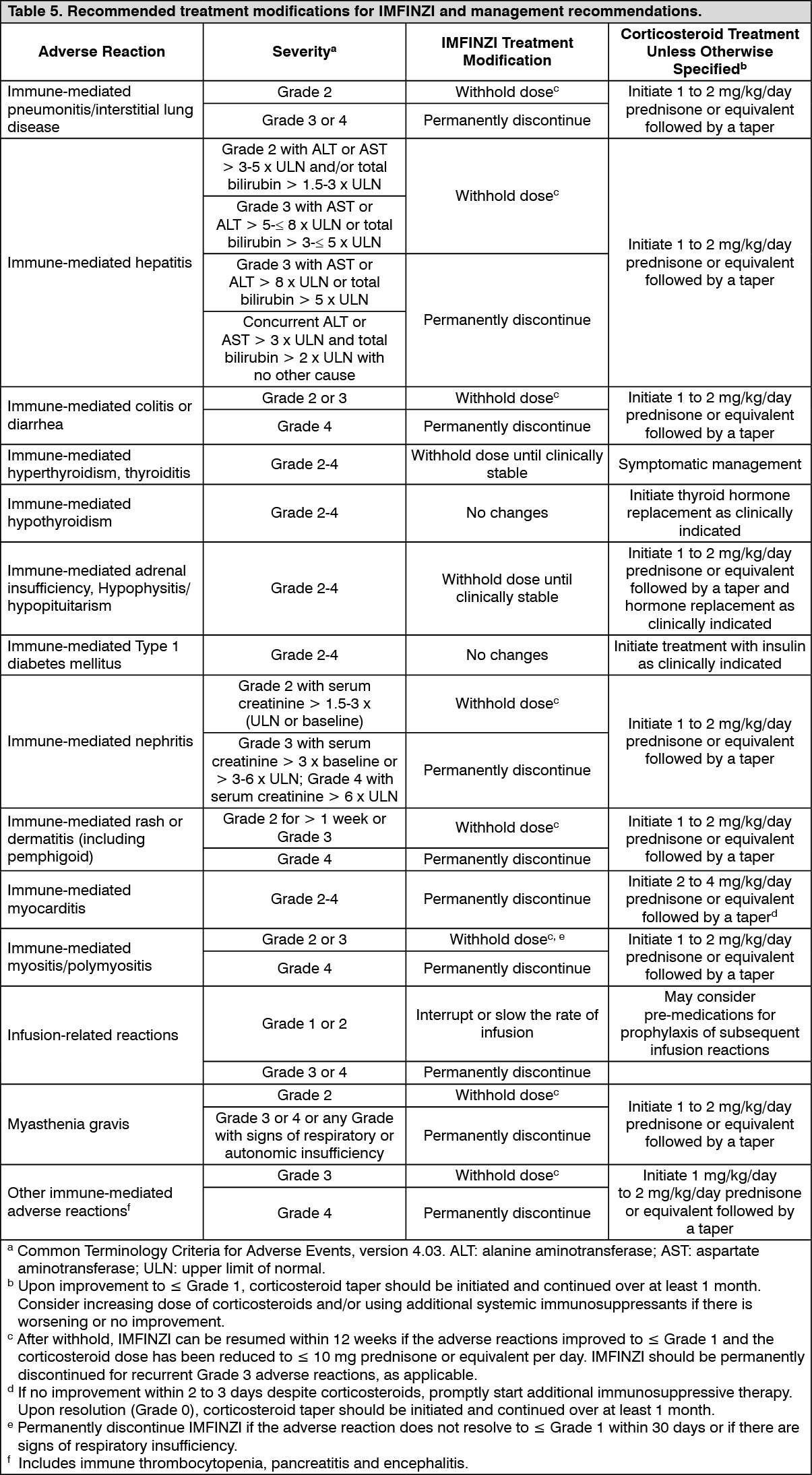 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image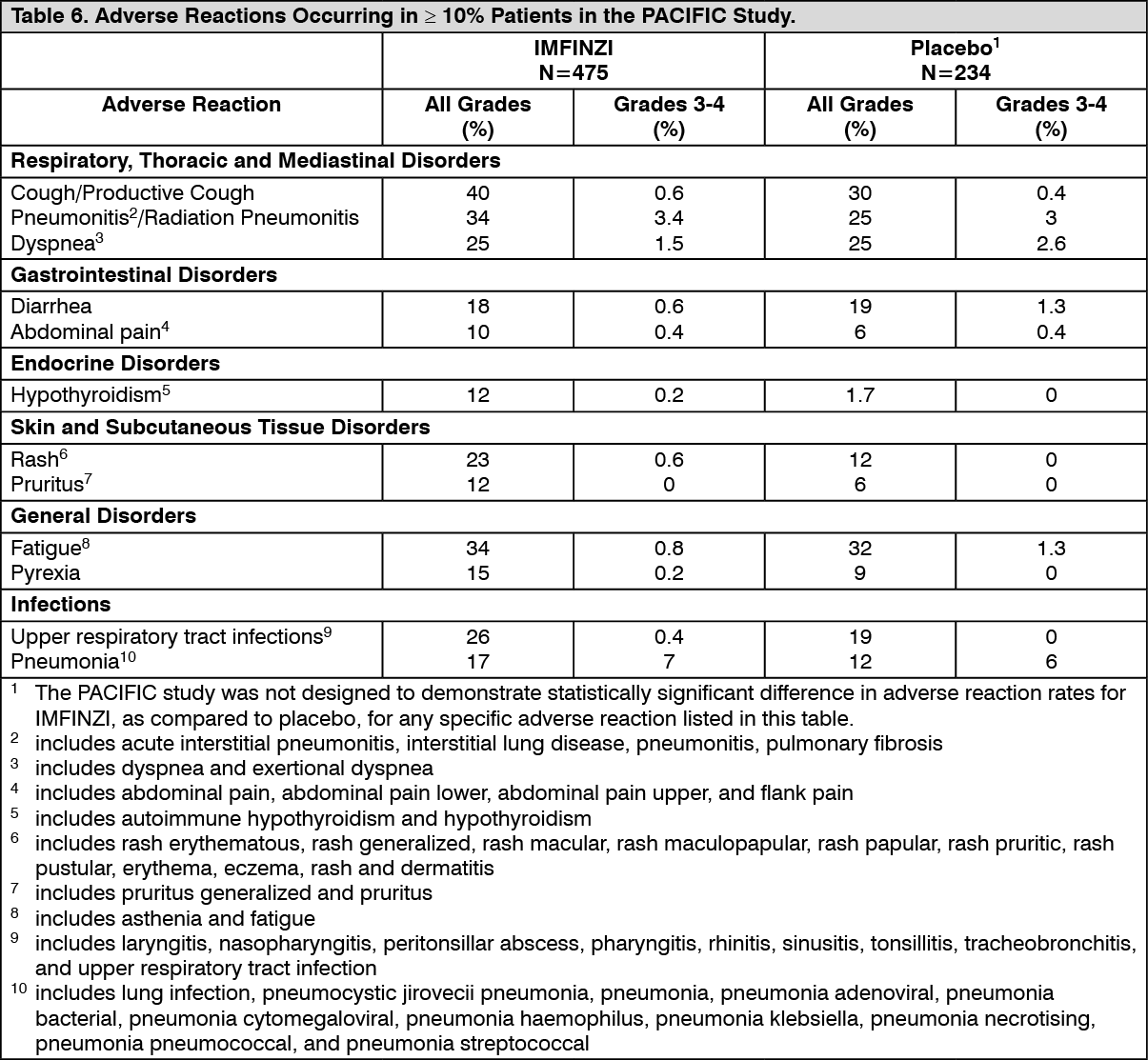 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image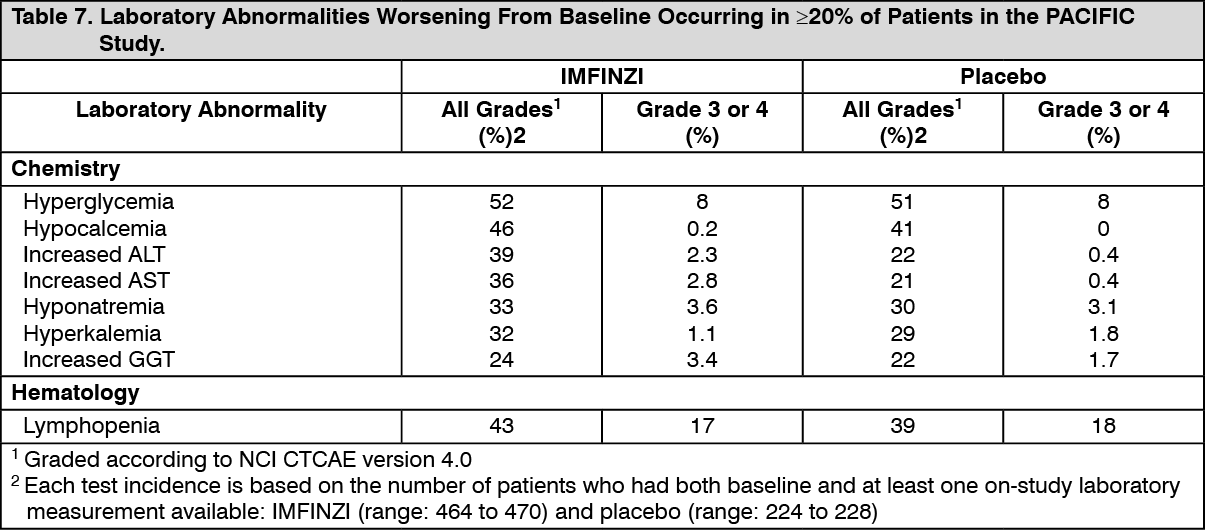 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image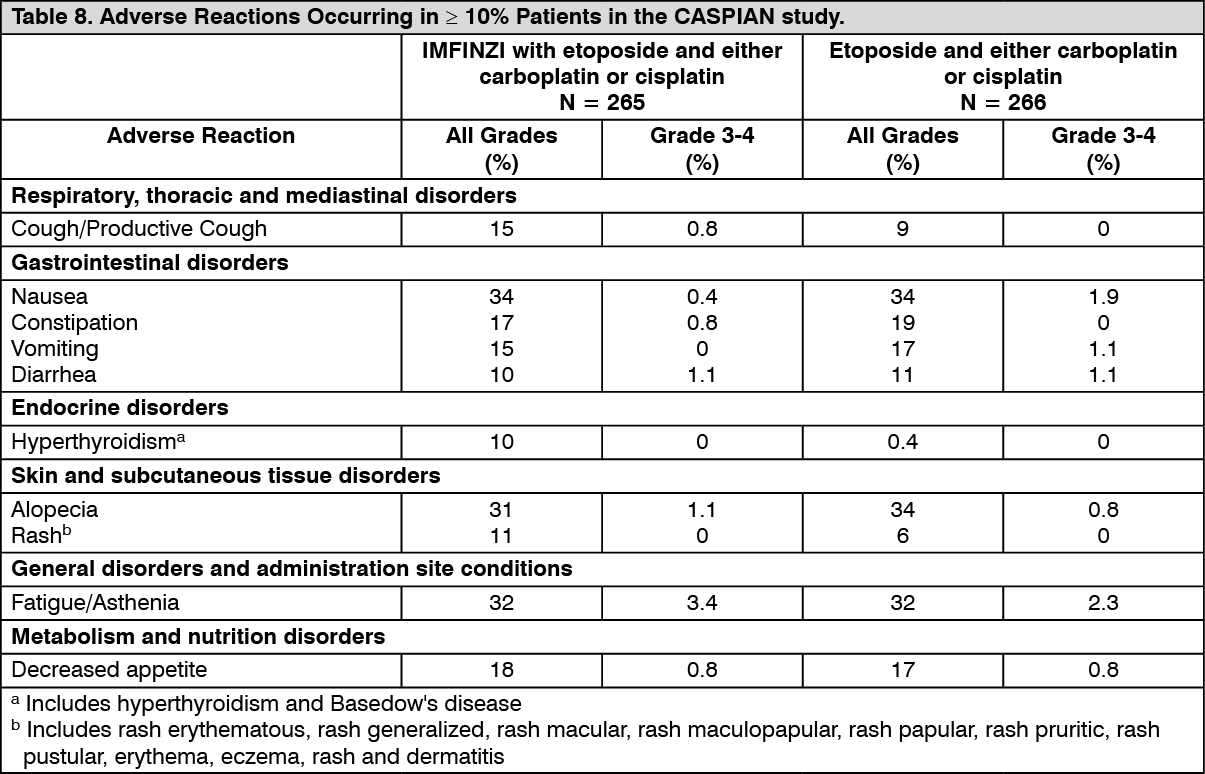 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image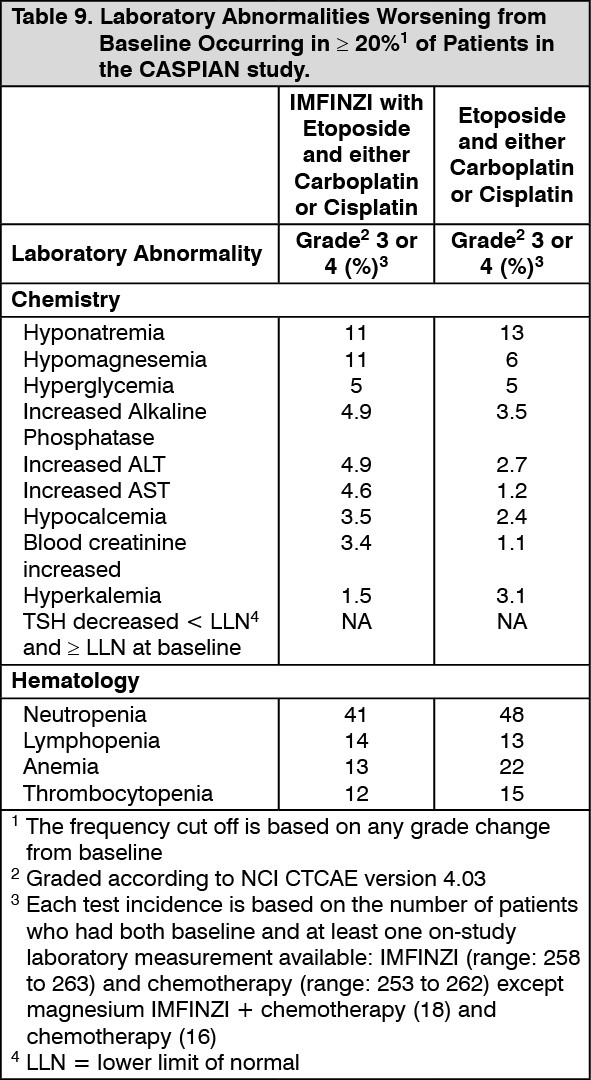 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image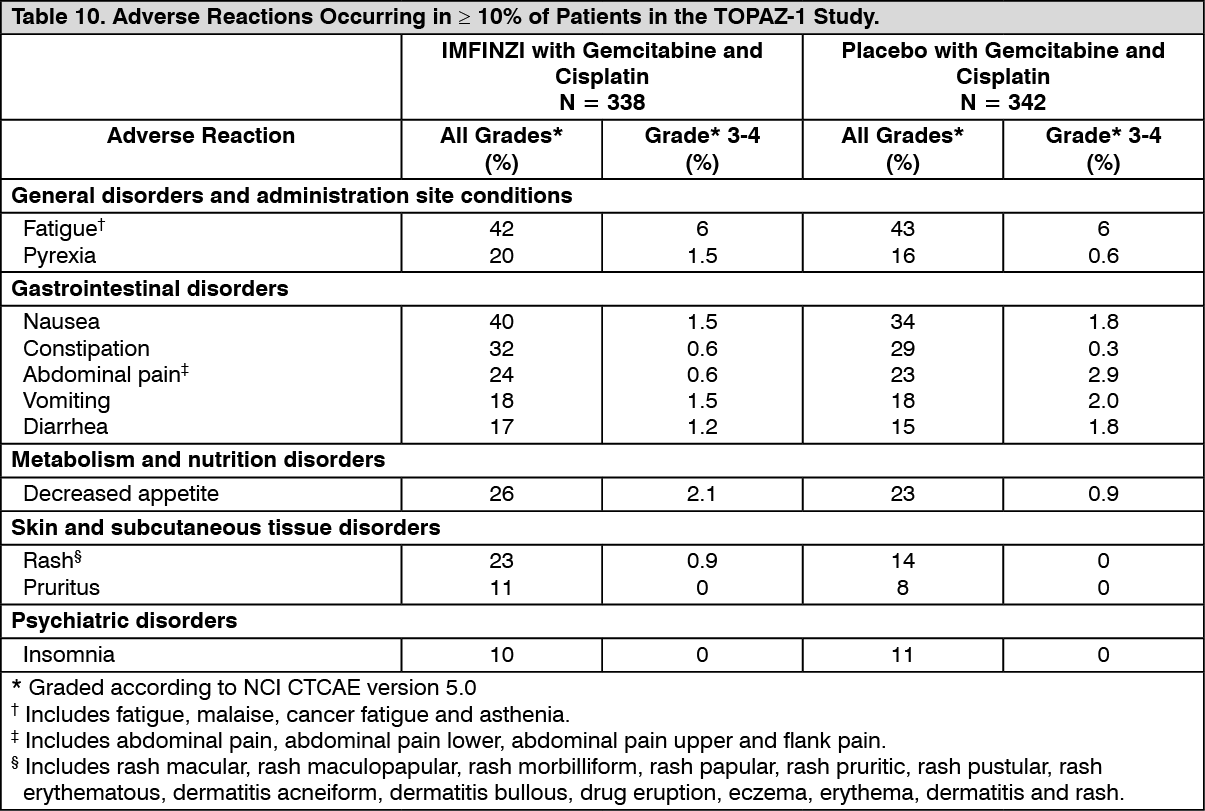 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image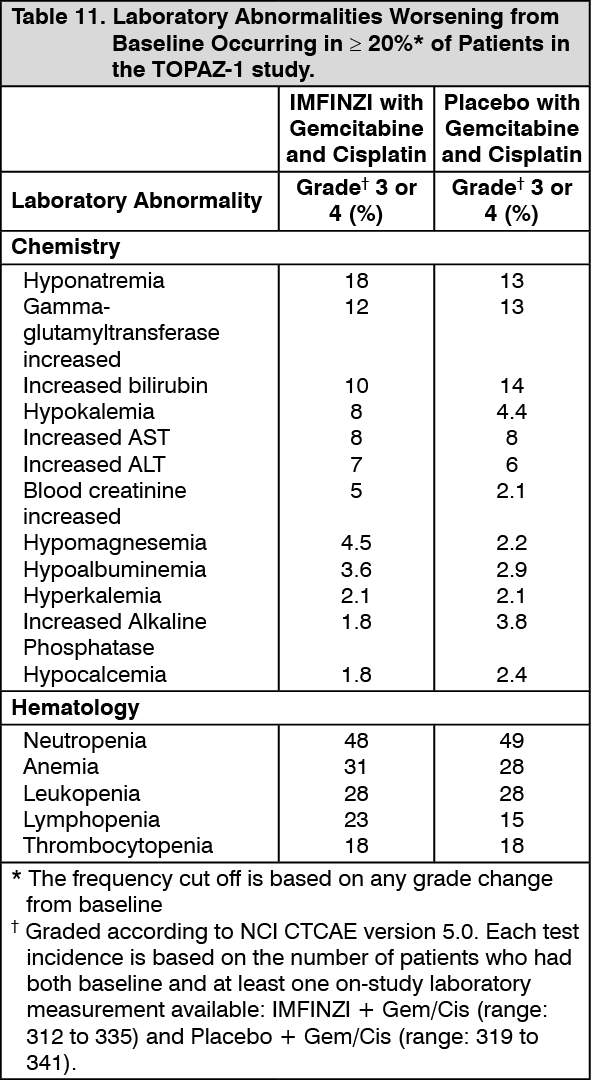 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 120 mg_2.4 mL25961993-e513-47d2-a771-afa20080fa59.GIF)
 500 mg_10 mLc573d05c-31de-422b-8dcd-afa20080fa67.GIF)

 Sign Out
Sign Out
