


 Sign Out
Sign Out

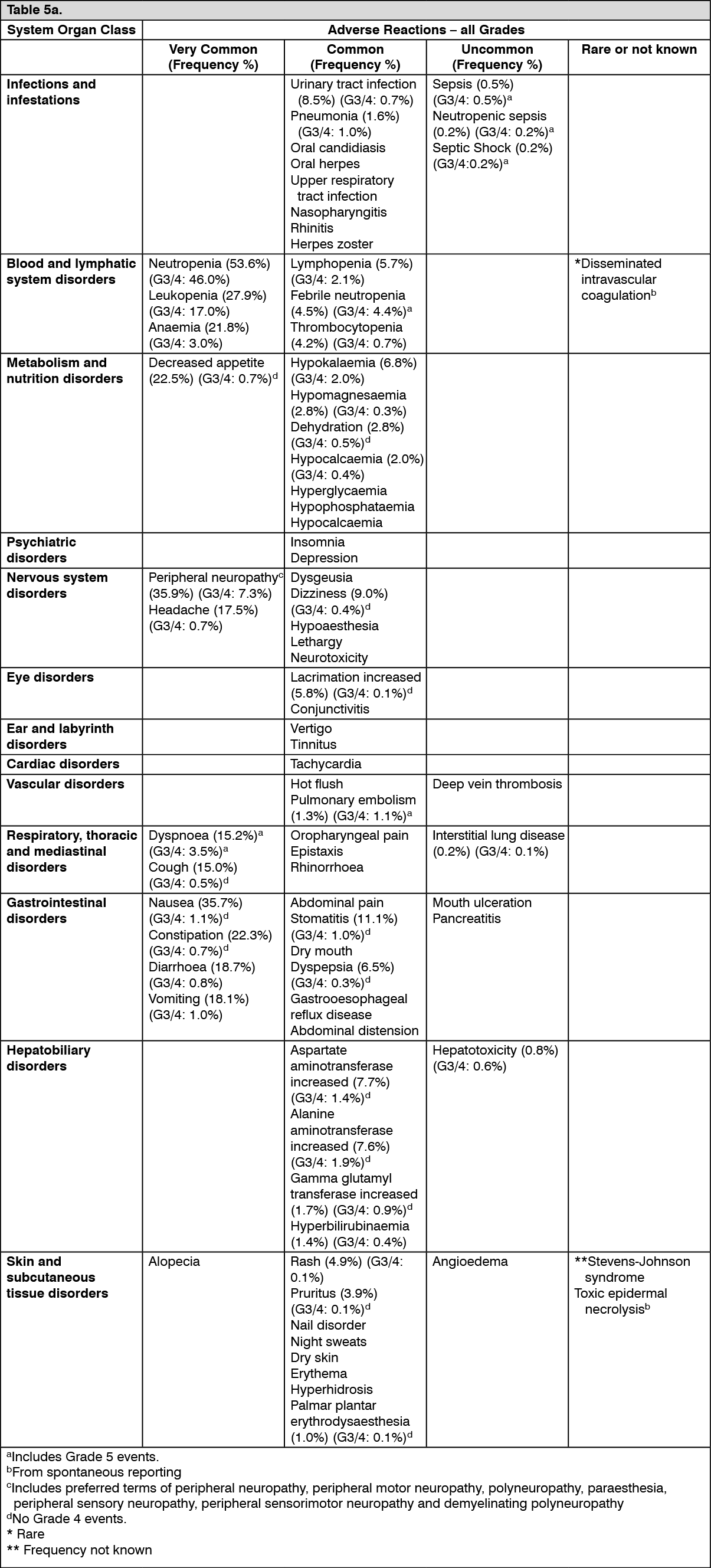
Tabulated list of adverse reactions: Unless otherwise noted, the table shows the incidence rates of adverse reactions observed in breast cancer and soft tissue sarcoma patients who received the recommended dose in Phase 2 and Phase 3 studies.
Frequency categories are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000).
Within each frequency grouping, undesirable effects are presented in order of decreasing frequency. Where Grade 3 or 4 reactions occurred, the actual total frequency and the frequency of Grade 3 or 4 reactions are given. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image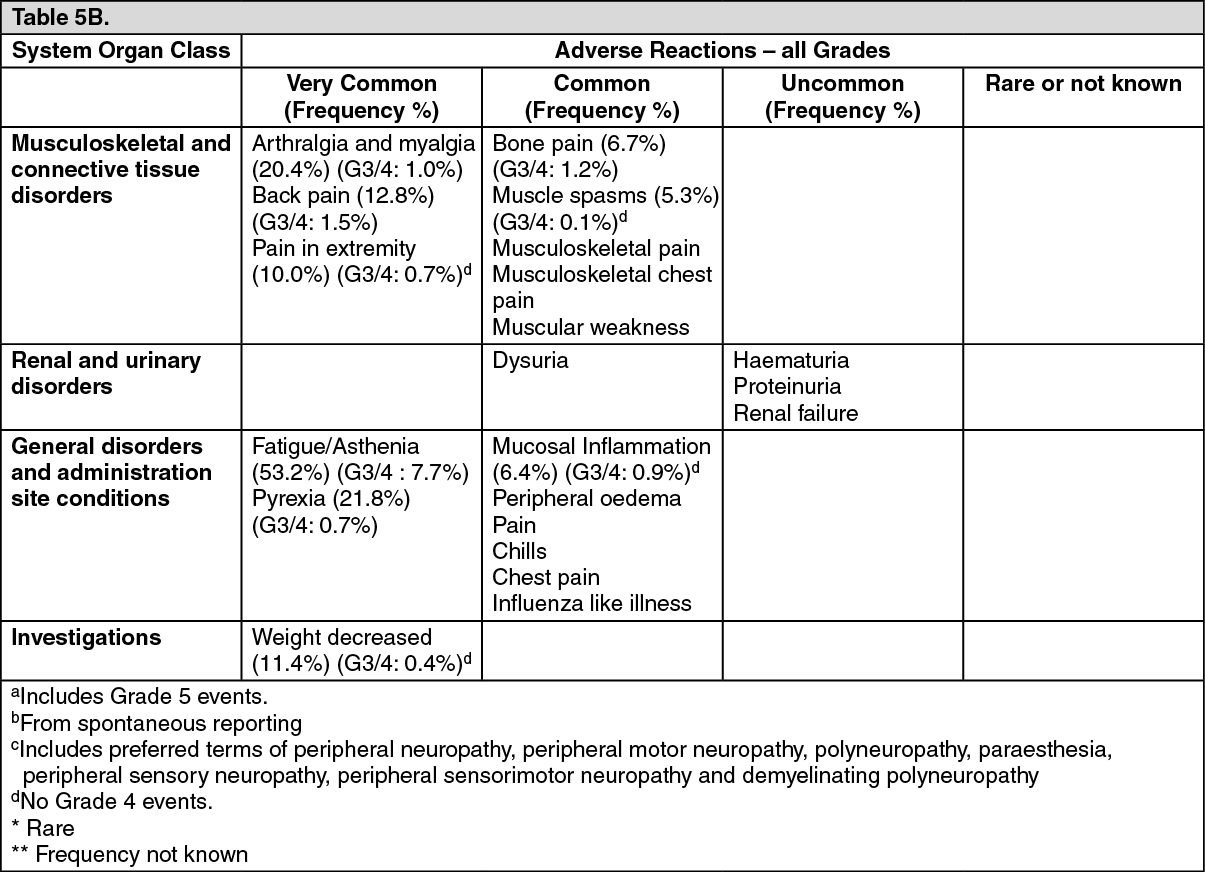 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOverall, the safety profiles in the breast cancer and soft tissue sarcoma patient populations were similar.
Description of selected adverse reactions: Neutropenia: The neutropenia observed was reversible and not cumulative; the mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (< 0.5 x 109/l) was 8 days.
Neutrophil counts of < 0.5 x 109/l that lasted for more than 7 days occurred in 13% of breast cancer patients treated with eribulin in the EMBRACE study.
Neutropenia was reported as a Treatment Emergent Adverse Event (TEAE) in 151/404 (37.4% for all grades) in the sarcoma population, compared with 902/1559 (57.9% for all grades) in the breast cancer population. The combined grouped TEAE and neutrophil laboratory abnormality frequencies were 307/404 (76.0%) and 1314/1559 (84.3%), respectively. The median duration of treatment was 12.0 weeks for sarcoma patients and 15.9 weeks for breast cancer patients.
Fatal cases of febrile neutropenia, neutropenic sepsis, sepsis and septic shock have been reported. Out of 1963 breast cancer and soft tissue sarcoma patients who received eribulin at the recommended dose in clinical trials there was one fatal event each of neutropenic sepsis (0.1%) and febrile neutropenia (0.1%). In addition there were 3 fatal events of sepsis (0.2%) and one of septic shock (0.1%).
Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines. 18% and 13% of eribulin treated patients received G-CSF in the two phase 3 breast cancer studies (Studies 305 and 301, respectively). In the phase 3 sarcoma study (Study 309), 26% of the eribulin treated patients received G-CSF.
Neutropenia resulted in discontinuation in < 1% of patients receiving eribulin.
Disseminated intravascular coagulation: Cases of disseminated intravascular coagulation have been reported, typically in association with neutropenia and/or sepsis.
Peripheral neuropathy: In the 1559 breast cancer patients the most common adverse reaction resulting in discontinuation of treatment with eribulin was peripheral neuropathy (3.4%). The median time to Grade 2 peripheral neuropathy was 12.6 weeks (post 4 cycles). Out of the 404 sarcoma patients, 2 patients discontinued treatment with eribulin due to peripheral neuropathy. The median time to Grade 2 peripheral neuropathy was 18.4 weeks.
Development of Grade 3 or 4 peripheral neuropathy occurred in 7.4% of breast cancer patients and 3.5% of sarcoma patients. In clinical trials, patients with pre-existing neuropathy were as likely to develop new or worsening symptoms as those who entered the study without the condition.
In breast cancer patients with pre-existing Grade 1 or 2 peripheral neuropathy the frequency of treatment-emergent Grade 3 peripheral neuropathy was 14%.
Hepatoxicity: In some patients with normal/abnormal liver enzymes prior treatment with eribulin, increased levels of liver enzymes have been reported with initiation of eribulin treatment. Such elevations appeared to have occurred early with eribulin treatment in cycle 1 - 2 for the majority of these patients and whilst thought likely to be a phenomenon of adaptation to eribulin treatment by the liver and not a sign of significant liver toxicity in most patients, hepatotoxicity has also been reported.
Special populations: Elderly population: Of the 1559 breast cancer patients treated with the recommended dose of eribulin, 283 patients (18.2%) were ≥ 65 years of age. In the 404 sarcoma patient population, 90 patients (22.3%) treated with eribulin were ≥ 65 years of age. The safety profile of eribulin in elderly patients (≥ 65 years of age) was similar to that of patients <65 years of age except for asthenia/fatigue which showed an increasing trend with age. No dose adjustments are recommended for the elderly population.
Patients with hepatic impairment: Patients with ALT or AST > 3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin > 1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia (see also Dosage & Administration and Pharmacology: Pharmacokinetics under Actions).
Paediatric population: Three open-label studies, Studies 113, 213 and 223, were conducted in paediatric patients with refractory or recurrent solid tumours and lymphomas, but excluding central nervous system (CNS) tumours (see Pharmacology: Pharmacodynamics under Actions).
The safety of eribulin monotherapy was evaluated in 43 paediatric patients who received up to 1.58 mg/m2 on Days 1 and 8 of a 21-day cycle (Studies 113 and 223). The safety of eribulin in combination with irinotecan was also evaluated in 40 paediatric patients who received eribulin 1.23 mg/m2 on Days 1 and 8 and irinotecan 20 or 40 mg/m2 on Days 1 to 5 of a 21-day cycle, or 100 or 125 mg/m2 on Days 1 and 8 of a 21-day cycle (Study 213).
In Study 113 (Phase 1), the most frequently reported adverse drug reactions were white blood cell count decreased, lymphocyte count decreased, anaemia and neutrophil count decreased.
In Study 213 (Phase 1/2), the most frequently reported adverse drug reactions were neutropenia (Phase 1) and diarrhoea and neutrophil count decreased (Phase 2).
In Study 223 (Phase 2), the most frequently reported adverse drug reactions were neutrophil count decreased, anaemia, and white blood cell count decreased.
The safety profile of eribulin as monotherapy or in combination with irinotecan hydrochloride in this paediatric population was consistent with the known safety profile of either study drug in the adult population.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
View ADR Monitoring Form