Amlodipine besylate, valsartan.
Exforge FCT are non-divisible and cannot be divided into equal doses.
Excipients/Inactive Ingredients: 5/80 mg: Cellulose microcrystalline; crospovidone; silica, colloidal anhydrous; magnesium stearate; hypromellose, macrogol 4000, talc, titanium dioxide (E171), iron oxide, yellow (E172).
5/160 mg: Cellulose microcrystalline; crospovidone; silica, colloidal anhydrous; magnesium stearate; hypromellose, macrogol 4000, talc, titanium dioxide (E171), iron oxide, yellow (E172).
10/160 mg: Cellulose microcrystalline; crospovidone; silica, colloidal anhydrous, magnesium stearate, hypromellose, macrogol 4000, talc, titanium dioxide (E171), iron oxide, yellow (E172), iron oxide, red (E172).
Pharmacotherapeutic group: angiotensin II antagonists, plain (valsartan) combinations with dihydropyridine derivatives (amlodipine). ATC code: C09DB01.
Pharmacology: Pharmacodynamics: Exforge combines two antihypertensive compounds with complementary mechanisms to control blood pressure in patients with essential hypertension: amlodipine belongs to the calcium antagonist class and valsartan to the angiotensin II (Ang II) antagonist class of medicines. The combination of these ingredients has an additive antihypertensive effect, reducing blood pressure to a greater degree than either component alone.
Amlodipine: The amlodipine component of Exforge inhibits the transmembrane entry of calcium ions into cardiac and vascular smooth muscle. The mechanism of the antihypertensive action of amlodipine is due to a direct relaxant effect on vascular smooth muscle, causing reductions in peripheral vascular resistance and blood pressure. Experimental data suggest that amlodipine binds to both dihydropyridine and nondihydropyridine binding sites. The contractile processes of cardiac muscle and vascular smooth muscle are dependent upon the movement of extracellular calcium ions into these cells through specific ion channels.
Following administration of therapeutic doses to patients with hypertension, amlodipine produces vasodilatation resulting in a reduction of supine and standing blood pressures. These decreases in blood pressure are not accompanied by a significant change in heart rate or plasma catecholamine levels with chronic dosing.
Plasma concentrations correlate with effect in both young and elderly patients.
In hypertensive patients with normal renal function, therapeutic doses of amlodipine resulted in a decrease in renal vascular resistance and an increase in glomerular filtration rate and effective renal plasma flow without change in filtration fraction or proteinuria.
As with other calcium channel blockers, haemodynamic measurements of cardiac function at rest and during exercise (or pacing) in patients with normal ventricular function treated with amlodipine have generally demonstrated a small increase in cardiac index without significant influence on dP/dt or on left ventricular end diastolic pressure or volume. In haemodynamic studies, amlodipine has not been associated with a negative inotropic effect when administered in the therapeutic dose range to intact animals and humans, even when co-administered with beta blockers to humans.
Amlodipine does not change sinoatrial nodal function or atrioventricular conduction in intact animals or humans. In clinical studies in which amlodipine was administered in combination with beta-blockers to patients with either hypertension or angina, no adverse experiences on electrocardiographic parameters were observed.
Amlodipine has demonstrated beneficial clinical effects in patients with chronic stable angina, vasospastic angina and angiographically documented coronary artery disease.
Valsartan: Valsartan is an orally active, potent, and specific angiotensin II receptor antagonist. It acts selectively on the AT1 receptor subtype, which is responsible for the known actions of angiotensin II. The increased plasma levels of angiotensin II following AT1 receptor blockade with valsartan may stimulate the unblocked AT2 receptor, which appears to counterbalance the effect of the AT1 receptor. Valsartan does not exhibit any partial agonist activity at the AT1 receptor and has much (about 20,000 fold) greater affinity for the AT1 receptor than for the AT2 receptor.
Valsartan does not inhibit ACE, also known as kininase II, which converts angiotensin I to angiotensin II and degrades bradykinin. Since there is no effect on ACE and no potentiation of bradykinin or substance P, angiotensin II antagonists are unlikely to be associated with cough. In clinical trials where valsartan was compared with an ACE inhibitor, the incidence of dry cough was significantly (P < 0.05) lower in patients treated with valsartan than in those treated with an ACE inhibitor (2.6% versus 7.9% respectively). In a clinical trial of patients with a history of dry cough during ACE inhibitor therapy, 19.5% of trial subjects receiving valsartan and 19.0% of those receiving a thiazide diuretic experienced cough compared to 68.5% of those treated with an ACE inhibitor (P < 0.05). Valsartan does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Administration of Valsartan to patients with hypertension results in reduction of blood pressure without affecting pulse rate.
In most patients, after administration of a single oral dose, onset of antihypertensive activity occurs within 2 hours, and the peak reduction of blood pressure is achieved within 4-6 hours. The antihypertensive effect persists over 24 hours after administration. During repeated administration, the maximum reduction in blood pressure with any dose is generally attained within 2-4 weeks and is sustained during long-term therapy. Abrupt withdrawal of Valsartan has not been associated with rebound hypertension or other adverse clinical events.
In patients with chronic heart failure (NYHA class II-IV), valsartan has been demonstrated to significantly reduce hospitalizations in patients with chronic heart failure (NYHA class II-IV). The benefits were greatest in patients not receiving either an ACE inhibitor or a beta blocker. In post-MI patients, valsartan has also been shown to reduce cardiovascular mortality in clinically stable patients with left ventricular failure or left ventricular dysfunction following myocardial infarction.
Clinical Studies: Over 1400 hypertensive patients received Exforge once daily in two placebo-controlled trials. The antihypertensive effect of a single dose of the combination persisted for 24 hours.
Exforge (amlodipine besylate/valsartan) was studied in 2 placebo-controlled trials in hypertensive patients with a diastolic blood pressure ≥ 95 mmHg and < 110 mmHg. In the first study (baseline blood pressure 153/99 mmHg), Exforge in doses of 5/80 mg, 5/160 mg and 5/320 mg reduced blood pressure 20-23/14-16 mmHg compared to 7/7 mmHg with placebo. In the second study (baseline blood pressure 157/99 mmHg), Exforge in doses of 10/160 mg and 10/320 mg reduced blood pressure 28/18-19 mmHg compared to 13/9 mmHg with placebo.
A multicenter, randomized, double-blind, active-controlled, parallel-group trial showed normalization of blood pressure (trough sitting diastolic BP < 90 mmHg at the end of the trial) in patients not adequately controlled on valsartan 160 mg in 75% of patients treated with amlodipine/valsartan 10 /160 mg and 62% of patients treated with amlodipine/valsartan 5/160 mg, compared to 53% of patients remaining on valsartan 160 mg. The addition of amlodipine 10 mg and 5 mg produced an additional reduction in systolic/diastolic blood pressure of 6.0/4.8 mmHg and 3.9/2.9 mmHg, respectively, compared to patients who remained on valsartan 160 mg only.
A multicenter, randomized, double-blind, active-controlled, parallel-group trial showed normalization of blood pressure (trough sitting diastolic BP < 90 mmHg at the end of the trial) in patients not adequately controlled on amlodipine 10 mg in 78% of patients treated with amlodipine/valsartan 10/160 mg, compared to 67% of patients remaining on amlodipine 10 mg. The addition of valsartan 160 mg produced an additional reduction in systolic/diastolic blood pressure of 2.9/2.1 mmHg compared to patients who remained on amlodipine 10 mg only.
Exforge was also studied in an active-controlled study of 130 hypertensive patients with diastolic blood pressure ≥ 110 mmHg and < 120 mmHg. In this study (baseline blood pressure 171/113 mmHg), an Exforge regimen of 5/160 mg titrated to 10/160 mg reduced sitting blood pressure by 36/29 mmHg as compared to 32/28 mmHg with a regimen of lisinopril/hydrochlorothiazide 10/12.5 mg titrated to 20/12.5 mg.
In other studies, the probability of achieving systolic or diastolic blood pressure control was greater with initial combination therapy than valsartan and amlodipine monotherapy at all levels of baseline blood pressure.
In two long-term follow-up studies the effect of Exforge was maintained for over one year. Abrupt withdrawal of Exforge has not been associated with a rapid increase in blood pressure.
In patients whose blood pressure is adequately controlled with amlodipine but who experience unacceptable edema, combination therapy may achieve similar blood pressure control with less edema.
Efficacy in subgroup populations: In double-blind controlled studies, age, gender, race and/or body mass index (≥30 kg/m2, <30 kg/m2) did not influence the response to Exforge.
Two double-blind, active-controlled studies were conducted in which Exforge was administered as initial therapy. In 1 study, a total of 572 black patients with moderate to severe hypertension were randomized to receive either the combination amlodipine/valsartan or amlodipine monotherapy for 12 weeks. The initial dose of amlodipine/valsartan was 5/160 mg for 2 weeks with forced titration to 10/160 mg for 2 weeks, followed by optional titration to 10/320 mg for 4 weeks and optional addition of HCTZ 12.5 mg for 4 weeks. The initial dose of amlodipine was 5 mg for 2 weeks with forced titration to 10 mg for 2 weeks, followed by optional titration to 10 mg for 4 weeks and optional addition of HCTZ 12.5 mg for 4 weeks. At the primary endpoint of 8 weeks, the treatment difference between amlodipine/valsartan and amlodipine was 6.7/2.8 mmHg.
In the other study of similar design, a total of 646 patients with moderate to severe hypertension (MSSBP of ≥160 mmHg and <200 mmHg) were randomized to receive either the combination amlodipine/valsartan or amlodipine monotherapy for 8 weeks. The initial dose of amlodipine/valsartan was 5/160 mg for 2 weeks with forced titration to 10/160 mg for 2 weeks, followed by the optional addition of HCTZ 12.5 mg for 4 weeks. The initial dose of amlodipine was 5 mg for 2 weeks with forced titration to 10 mg for 2 weeks, followed by the optional addition of HCTZ 12.5 mg for 4 weeks. At the primary endpoint of 4 weeks, the treatment difference between amlodipine/valsartan and amlodipine was 6.6/3.9 mmHg.
EXCITE (EXperienCe of amlodIpine and valsarTan in hypErtension) Study: In an open, uncontrolled study, 9,794 hypertensive patients across 13 countries in the Middle East and Asia were treated according to routine clinical practice and prospectively observed for 26 weeks. A total of 8,603 were prescribed amlodipine/valsartan and 1,191 prescribed amlodipine/valsartan/hydrochlorothiazide. Among these, 15.5% were elderly, 32.5% were obese, 31.3% had diabetes, and 9.8% had isolated systolic hypertension. Both amlodipine/valsartan and amlodipine/valsartan/hydrochlorothiazide single-pill combinations, respectively, were associated with clinically relevant and significant mean sitting systolic/diastolic BP reductions in the overall population (-31.0/-16.6 mmHg and -36.6/-17.8 mmHg, respectively. These results were consistent regardless of age, body mass index, and diabetic status. Similarly, clinically relevant and significant systolic BP reductions were observed in patients with isolated systolic hypertension (-25.5 mmHg and -30.2 mmHg, respectively).
Asian studies (in Chinese and Taiwanese patients): Three Asian studies including more than 12,000 patients with hypertension, mostly of Chinese origin, have shown similar efficacy and safety of Exforge compared to the global registration studies in mixed, but predominantly Caucasian patients.
In a multicenter, open-label, prospective, observational study in China, with 11,422 enrolled hypertensive patients including 16.5% with diabetes and 3.1% with renal impairment, Exforge provided clinically meaningful and statistically significant reductions in mean sitting systolic blood pressure (MSSBP) and mean sitting diastolic BP (MSDBP) (mean reductions of 27.1 and 15.3 mmHg, respectively; p <0.0001) after 8 weeks of treatment. The BP goal originally defined as <130/80 mmHg for patients with diabetes or renal impairment, and <140/90 mmHg for all other patients was achieved by 66.1% of patients at Week 8.The unified defined BP goal (<140/90 mmHg for all patients) was achieved by 76.8% of patients at Week 8.
In a multicenter, randomized, open-label, active-control, parallel group study in China of Exforge compared with nifedipine gastrointestinal therapeutic system (GITS), with 564 hypertensive patients including 9.2% (Exforge arm) and 9.7% (nifedipine GITS arm) of patients with diabetes, Exforge resulted in 5.8 and 4.0 mmHg greater mean reductions in MSSBP and MSDBP (p <0.0001) compared with nifedipine GITS (mean reductions of 16.6 and 8.6 mmHg with Exforge vs. 10.8 and 4.6 mmHg with nifedipine GITS) after 12 weeks of treatment. The percentage of patients achieving the BP target (<140/90 or <130/80 mmHg in the absence or presence of diabetes mellitus, respectively) was significantly higher with Exforge (79.0%) vs. nifedipine GITS (57.4%; p<0.0001).
In a multicenter, open-label, prospective, observational study in Taiwan, with 1,029 enrolled hypertensive patients including 39.8% with diabetes, Exforge (administered alone or as add-on therapy) resulted in mean reductions of 12.5 and 6.5 mmHg in MSSBP and MSDBP, respectively, after 12 weeks of treatment. Overall, 48.3% patients receiving Exforge achieved the desired therapeutic BP goal.
Pharmacokinetics: Linearity: Valsartan and amlodipine exhibit linear pharmacokinetics.
Amlodipine: Absorption: After oral administration of therapeutic doses of amlodipine alone, peak plasma concentrations of amlodipine are reached in 6 to 12 hours. Absolute bioavailability has been calculated as between 64% and 80%. Amlodipine bioavailability is unaffected by food ingestion.
Distribution: Volume of distribution is approximately 21 L/kg. In vitro studies with amlodipine have shown that approximately 97.5% of circulating drug is bound to plasma proteins. Amlodipine crosses the placenta and is excreted into breast milk.
Biotransformation: Amlodipine is extensively (approximately 90%) metabolized in the liver to inactive metabolites.
Elimination: Amlodipine elimination from plasma is biphasic with a terminal elimination half-life of approximately 30 to 50 hours. Steady-state plasma levels are reached after continuous administration for 7-8 days. Ten per cent of original amlodipine and 60% of amlodipine metabolites are excreted in urine.
Valsartan: Absorption: Following oral administration of valsartan alone, peak plasma concentrations of valsartan are reached in 2 -4 hours. Mean absolute bioavailability is 23%. Food decreases the exposure (as measured by AUC) to valsartan by about 40% and peak plasma concentration (Cmax) by about 50%, although from about 8 h post dosing plasma valsartan concentrations are similar for the fed and fasted group. This reduction in AUC, however, is not accompanied by a clinically significant reduction in therapeutic effect, and valsartan can therefore be given either with or without food.
Distribution: The steady-state volume of distribution of valsartan after intravenous administration is about 17 liters indicating that valsartan is not distributed into tissues extensively. Valsartan is highly bound to serum proteins (94-97%), mainly serum albumin.
Biotransformation: Valsartan is not transformed to a high extent as only about 20% of dose is recovered as metabolites. A hydroxy metabolite has been identified in plasma at low concentrations (less than 10 % of the valsartan AUC). This metabolite is pharmacologically inactive.
Elimination: Valsartan shows multiexponential decay kinetics (1/2α <1h and t 1/2 β about 9 h). Valsartan is primarily eliminated unchanged in faeces (about 83% of dose) and urine (about 13% of dose) mainly as unchanged drug. Following intravenous administration, plasma clearance of valsartan is about 2 L/h and its renal clearance is 0.62 L/h (about 30% of total clearance). The half-life of valsartan is 6 hours.
Valsartan/Amlodipine: Following oral administration of Exforge peak plasma concentrations of valsartan and amlodipine are reached in 3 and 6-8 hours, respectively. The rate and extent of absorption of Exforge are equivalent to the bioavailability of valsartan and amlodipine when administered as individual tablets.
Special populations: Geriatric patients: The time to reach peak plasma concentrations of amlodipine is similar in elderly and younger subjects. Amlodipine clearance tends to be decreased with resulting increases in AUC and elimination half life in elderly patients.
Systemic exposure to valsartan is slightly elevated in the elderly as compared to the young, but this has not been shown to have any clinical significance.
Renal impairment: The pharmacokinetics of amlodipine is not significantly influenced by renal impairment. There is no apparent correlation between renal function (measured by creatinine clearance) and exposure (measured by AUC) to valsartan in patients with different degrees of renal impairment. Patients with mild to moderate renal impairment may therefore receive the usual initial dose (see DOSAGE & ADMINISTRATION and also PRECAUTIONS).
Hepatic impairment: Patients with hepatic insufficiency have decreased clearance of amlodipine with resulting increase in AUC of approximately 40-60%. In a small number of patients with mild to moderate hepatic impairment given single doses of 5 mg, amlodipine half-life has been prolonged. Worsening of liver function test values may occur.
About 70% of the absorbed valsartan dose is excreted in the bile, mainly as unchanged compound. The AUC with valsartan has been observed to approximately double in patients with mild or moderate hepatic impairment including patients with biliary obstructive disorders (see PRECAUTIONS). There are no data available on the use of valsartan in patients with severe hepatic dysfunction (see CONTRAINDICATIONS).
Care should be exercised in patients with liver disease (see DOSAGE & ADMINISTRATION and PRECAUTIONS).
Toxicology: Non-Clinical Safety Data: Amlodipine:Valsartan: In a variety of preclinical safety studies conducted in several animal species with amlodipine:valsartan, there were no findings that would exclude the use of therapeutic doses of amlodipine: valsartan in humans. Animal studies lasting 13 weeks have been conducted with amlodipine:valsartan combination in rats and marmosets, as well as studies in rats to investigate embryofetal development toxicity.
In a 13-week oral toxicity study in rats, amlodipine/valsartan-related inflammation of the glandular stomach was observed in males at doses ≥3/48 mg/kg/day and in female at doses ≥7.5/120 mg/kg/day. No such effects have been observed in the 13-week marmoset study at any dose, although inflammation of the large intestine was observed in the high-dose marmosets only (no effects at dose ≤5/80 mg/kg/day). The gastrointestinal adverse effects observed in clinical trials with Exforge were no more frequent with the combination than with the respective monotherapies.
The combination amlodipine:valsartan was not tested for mutagenicity, clastogenicity, reproductive performance or carcinogenicity as there was no evidence for any interaction between the two compounds.
Amlodipine: Safety data for amlodipine are well established both clinically and non-clinically. No relevant findings were observed in carcinogenicity studies, mutagenicity studies.
There was no effect on the fertility of rats treated with amlodipine (males for 64 days and females 14 days prior to mating) at doses up to 10 mg/kg/day (8 times the maximum recommended human dose of 10 mg on a mg/m2 basis, based on patient weight of 50 kg).
Amlodipine has been tested individually for mutagenicity, clastogenicity, reproductive performance and carcinogenicity with negative results.
Valsartan: Preclinical data revealed no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity, carcinogenic potential and effects on fertility.
Safety pharmacology and Long term toxicity: In a variety of preclinical safety studies conducted in several animal species, there were no findings that would exclude the use of therapeutic doses of valsartan in humans.
In preclinical safety studies, high doses of valsartan (200 to 600 mg/kg/day body weight) caused in rats a reduction of red blood cell parameters (erythrocytes, hemoglobin, hematocrit) and evidence of changes in renal hemodynamics (slightly raised blood urea nitrogen, and renal tubular hyperplasia and basophilia in males). These doses in rats (200 and 600 mg/kg/day) are approximately 6 and 18 times the maximum recommended human dose on a mg/m2 basis (calculations assume an oral dose of 320 mg/day and a 60-kg patient). In marmosets at comparable doses, the changes were similar though more severe, particularly in the kidney where the changes developed to a nephropathy including raised blood urea nitrogen and creatinine. Hypertrophy of the renal juxtaglomerular cells was also seen in both species. All changes were considered to be caused by the pharmacological action of valsartan which produces prolonged hypotension, particularly in marmosets. For therapeutic doses of valsartan in humans, the hypertrophy of the renal juxtaglomerular cells does not seem to have any relevance.
Reproductive toxicity: In a rat fertility study, Valsartan had no adverse effects on the reproductive performance of male or female rats at oral doses up to 200 mg/kg/day, approximately 18 times the maximum recommended human dose on a mg/m2 basis (calculations assume an oral dose of 320 mg/day and a 60-kg patient).
Mutagenicity: Valsartan was devoid of mutagenic potential at either the gene or chromosome level when investigated in various standard in vitro and in vivo genotoxicity studies.
Carcinogenicity: There was no evidence of carcinogenicity when valsartan was administered in the diet to mice and rats for 2 years at doses up to 160 and 200 mg/kg/day, respectively.
Treatment of essential hypertension.
Exforge is indicated in patients whose blood pressure is not adequately controlled by monotherapy.
Exforge is indicated for the initial treatment of hypertension. The choice of Exforge for initial treatment should be based on an assessment of the potential benefits and risks.
Dosage regimen: General target population: A patient whose blood pressure is not adequately controlled on monotherapy may be switched to combination therapy with Exforge. The recommended dose is one tablet per day (the 3 strengths are listed under DESCRIPTION). When clinically appropriate direct change from monotherapy to the fixed-dose combination may be considered.
For convenience, patients receiving valsartan and amlodipine from separate tablets may be switched to Exforge containing the same component doses.
For initial therapy the usual starting dose is Exforge 5mg/80 mg once daily. The dosage can be increased after 1 to 2 weeks of therapy to a maximum of 10 mg/320 mg per day as needed to control blood pressure. Exforge is not recommended as initial therapy in patients with intravascular volume depletion (see Precautions).
The maximum dose is 10 mg/320 mg per day.
Both amlodipine and valsartan monotherapy can be taken with or without food. It is recommended to take Exforge with some water.
Special populations: Geriatric patients (aged 65 years or above): Since both components of the combination are equally well tolerated when used at similar doses in elderly (aged 65 years or above) or younger patients, no dose adjustment of the starting dose is required. Starting with the lowest available dose of amlodipine should be considered. The lowest strength of Exforge contains 5 mg of amlodipine (see PHARMACOLOGY under Actions).
Pediatric patients (below 18 years): Exforge is not recommended for use in patients aged below 18 years due to a lack of data on safety and efficacy.
Renal impairment: No dosage adjustment is required for patients with mild to moderate renal impairment. Caution is required if severe renal impairment occur.
Hepatic impairment: Liver function should be monitored in patients with mild to moderate hepatic impairment. The daily dose of Exforge should not exceed 5/80mg in patients with mild to moderate hepatic impairment without cholestasis. Exforge is contraindicated in severe hepatic impairment. Starting with the lowest available dose of amlodipine should be considered. The lowest strength of Exforge contains 5 mg of amlodipine.
There is no experience of overdosage with Exforge yet. The major symptom of overdosage with valsartan is possibly pronounced hypotension with dizziness. Overdosage with amlodipine may result in excessive peripheral vasodilatation and possibly reflex tachycardia. Marked and potentially prolonged systemic hypotension up to and including shock with fatal outcome have been reported.
Clinically significant hypotension due to amlodipine overdosage calls for active cardiovascular support including frequent monitoring of cardiac and respiratory function, elevation of extremities, and attention to circulating fluid volume and urine output.
A vasoconstrictor may be helpful in restoring vascular tone and blood pressure, provided that there is no contraindication to its use.
If the ingestion is recent, induction of vomiting or gastric lavage may be considered.
Administration of activated charcoal to healthy volunteers immediately or up to two hours after ingestion of amlodipine has been shown to significantly decrease amlodipine absorption.
Intravenous calcium gluconate may be beneficial in reversing the effects of calcium channel blockade.
Both valsartan and amlodipine are unlikely to be removed by haemodialysis.
Known hypersensitivity to the amlodipine, valsartan or to any of the excipients.
Pregnancy (see USE IN PREGNANCY & LACTATION).
Severe hepatic impairment; biliary cirrhosis and cholestasis.
Concomitant use of angiotensin receptor antagonists (ARBs) - including valsartan - or of angiotensin-converting enzyme inhibitors (ACEIs) with aliskiren in patients with Type 2 diabetes (see Dual blockade of the RAS under INTERACTIONS).
Patients with sodium- and/or volume depletion: Excessive hypotension was seen in 0.4% of patients with uncomplicated hypertension treated with Exforge in placebo-controlled studies. In patients with an activated renin-angiotensin system (such as volume- and/or salt-depleted patients receiving high doses of diuretics) who are receiving angiotensin receptor blockers, symptomatic hypotension may occur. Correction of this condition prior to administration of Exforge or close medical supervision at the start of treatment is recommended.
If hypotension occurs with Exforge, the patient should be placed in the supine position and, if necessary, given an i.v. infusion of normal saline. Treatment can be continued once blood pressure has been stabilized.
Hyperkalemia: Concomitant use with potassium supplements, potassium sparing diuretics, salt substitutes containing potassium, or other drugs that may increase potassium levels (heparin, etc.) should be used with caution and with frequent monitoring of potassium.
Patients with renal artery stenosis: Exforge should be used with caution to treat hypertension in patients with unilateral or bilateral renal artery stenosis, stenosis to a solitary kidney since blood urea and serum creatinine may increase in such patients.
Patients with renal impairment: No data is available for severe cases (creatinine clearance < 10 mL/min.) and caution is therefore advised. No dosage adjustment of Exforge is required for patients with mild to moderate renal impairment.
The use of ARBs - including valsartan - or of ACEIs with aliskiren should be avoided in patients with severe renal impairment (GFR < 30 mL/min) (see Dual blockade of the RAS under INTERACTIONS,).
Patients with kidney transplantation: To date there is no experience of the safe use of Exforge in patients who have had a recent kidney transplantation.
Patients with hepatic impairment: Valsartan is mostly eliminated unchanged via the bile whereas amlodipine is extensively metabolized by the liver. In patient with mild to moderate hepatic impairment without cholestasis, Exforge should be used with caution (see Pharmacology: PHARMACOKINETICS under Actions) and careful monitoring of liver function tests should be performed. The daily dose of Exforge should not exceed 5/80mg. Patients with severe hepatic impairment, biliary cirrhosis or cholestasis should not take Exforge (see CONTRAINDICATIONS).
Angioedema: Angioedema, including swelling of the larynx and glottis, causing airway obstruction and/or swelling of the face, lips, pharynx, and/or tongue has been reported in patients treated with valsartan; some of these patients previously experienced angioedema with other drugs including ACE inhibitors. Exforge should be immediately discontinued in patients who develop angioedema, and Exforge should not be re-administered.
Patients with heart failure/post-myocardial infarction: In general, calcium channel blockers including amlodipine should be used with caution in patients with serious congestive heart failure (New York Heart Association (NYHA) functional class III-IV).
In patients whose renal function may depend on the activity of the renin-angiotensin-aldosterone system (e.g. patients with severe congestive heart failure), treatment with angiotensin converting enzyme inhibitors or angiotensin receptor antagonists has been associated with oliguria and/or progressive azotemia, and in rare cases with acute renal failure and/or death. Evaluation of patients with heart failure or post-myocardial infarction should always include assessment of renal function.
Patients with acute myocardial infarction: Worsening angina pectoris and acute myocardial infarction can develop after starting or increasing the dose of amlodipine, particularly in patients with severe obstructive coronary artery disease.
Patients with aortic and mitral valve stenosis, obstructive hypertrophic cardiomyopathy: As with all other vasodilators, special caution is required when using amlodipine in patients suffering from aortic or mitral stenosis, or obstructive hypertrophic cardiomyopathy.
Dual Blockade of the Renin-Angiotensin System (RAS): Caution is required while co-administering ARBs, including valsartan, with other agents blocking the RAS such as ACEIs or aliskiren (see Dual blockade of the RAS under INTERACTIONS).
Pregnancy: Risk summary: As for any drug that acts directly on the RAAS, Exforge must not be used during pregnancy (see CONTRAINDICATIONS). Due to the mechanism of action of angiotensin II antagonists, a risk to the foetus cannot be excluded. Administration of angiotensin converting enzyme (ACE) inhibitors (a specific class of drugs acting on the renin-angiotensin-aldosterone system, RAAS) to pregnant women during the second and third trimesters has been reported to cause injury and death to the developing foetus. In addition, in retrospective data, first trimester use of ACE inhibitors has been associated with a potential risk of birth defects. There have been reports of spontaneous abortion, oligohydramnios and newborn renal dysfunction when pregnant women have inadvertently taken valsartan.
There are no adequate clinical data with amlodipine in pregnant women. Animal studies with amlodipine have shown reproductive toxicity at dose 8 times the maximum recommended dose of 10 mg (see Pharmacology: Toxicology: NON-CLINICAL SAFETY DATA under Actions). The potential risk to humans is unknown.
If pregnancy is detected during therapy, Exforge must be discontinued as soon as possible (see ANIMAL DATA as follows).
Clinical considerations: Disease-associated maternal and/or embryo/fetal risk: Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death.
Fetal/Neonatal Risk: Oligohydramnios in pregnant women who use drugs affecting the renin-angiotensin system in the second and third trimesters of pregnancy can result in the following: reduced fetal renal function leading to anuria and renal failure, fetal lung hypoplasia, skeletal deformations, including skull hypoplasia, hypotension and death.
In case of accidental exposure to ARB therapy, appropriate fetal monitoring should be considered.
Infants whose mothers have taken ARB therapy in the first trimester, should be closely observed for hypotension.
Animal data: Valsartan and amlodipine: In an oral embryo-fetal development study in rats with dose levels of 5:80 mg/kg/day, amlodipine:valsartan, 10:160 mg/kg/day amlodipine:valsartan, and 20:320 mg/kg/day amlodipine:valsartan, treatment-related maternal and fetal effects (developmental delays and alterations noted in the presence of significant maternal toxicity) were noted with the high dose combination. The no-observed-adverse-effect level (NOAEL) for embryo-fetal effects was 10:160 mg/kg/day amlodipine:valsartan. These doses are, respectively, 4.3 and 2.7 times the systemic exposure in humans receiving the MRHD (10/320 mg/60 kg).
Valsartan: In embryofetal development studies in mice, rats and rabbits, fetotoxicity was observed in association with maternal toxicity in rats at valsartan doses of 600 mg/kg/day approximately 6 times the maximum recommended human dose on a mg/m2 basis (calculations assume an oral dose of 320 mg/day and a 60-kg patient) and in rabbits at doses of 10 mg/kg/day approximately 0.6 times the maximum recommended human dose on a mg/m2 basis (calculations assume an oral dose of 320 mg/day and a 60-kg patient). There was no evidence of maternal toxicity or fetotoxicity in mice up to a dose level of 600 mg/kg/day approximately 9 times the maximum recommended human dose on a mg/m2 basis (calculations assume an oral dose of 320 mg/day and a 60-kg patient).
Amlodipine: No evidence of teratogenicity or embryo/fetal toxicity was found when pregnant rats and rabbits were treated orally with amlodipine maleate at doses up to 10 mg amlodipine/kg/day during their respective periods of major organogenesis. However, litter size was significantly decreased (by about 50%) and the number of intrauterine deaths was significantly increased (about 5-fold). Amlodipine has been shown to prolong both the gestation period and the duration of labor in rats at this dose.
Lactation: It is not known whether valsartan is excreted in human milk. It is reported that amlodipine is excreted in human milk. The proportion of the maternal dose received by the infant has been estimated with an interquartile range of 3 - 7%, with a maximum of 15%. The effect of amlodipine on infants is unknown. Valsartan was excreted in the milk of lactating rats. It is therefore not advisable for women who are breast-feeding to use Exforge.
Females and males of reproductive potential: As for any drug that acts directly on the RAAS, Exforge must not be used in women planning to become pregnant. Healthcare professionals prescribing any agents acting on the RAAS should counsel women of childbearing potential about the potential risk of these agents during pregnancy.
Infertility: There is no information on the effects of amlodipine or valsartan on human fertility. Studies in rats did not show any effects of amlodipine or valsartan on fertility (see Pharmacology: Toxicology: NON-CLINICAL SAFETY DATA under Actions).
The safety of Exforge has been evaluated in five controlled clinical studies with 5,175 patients, 2,613 of whom received valsartan in combination with amlodipine.
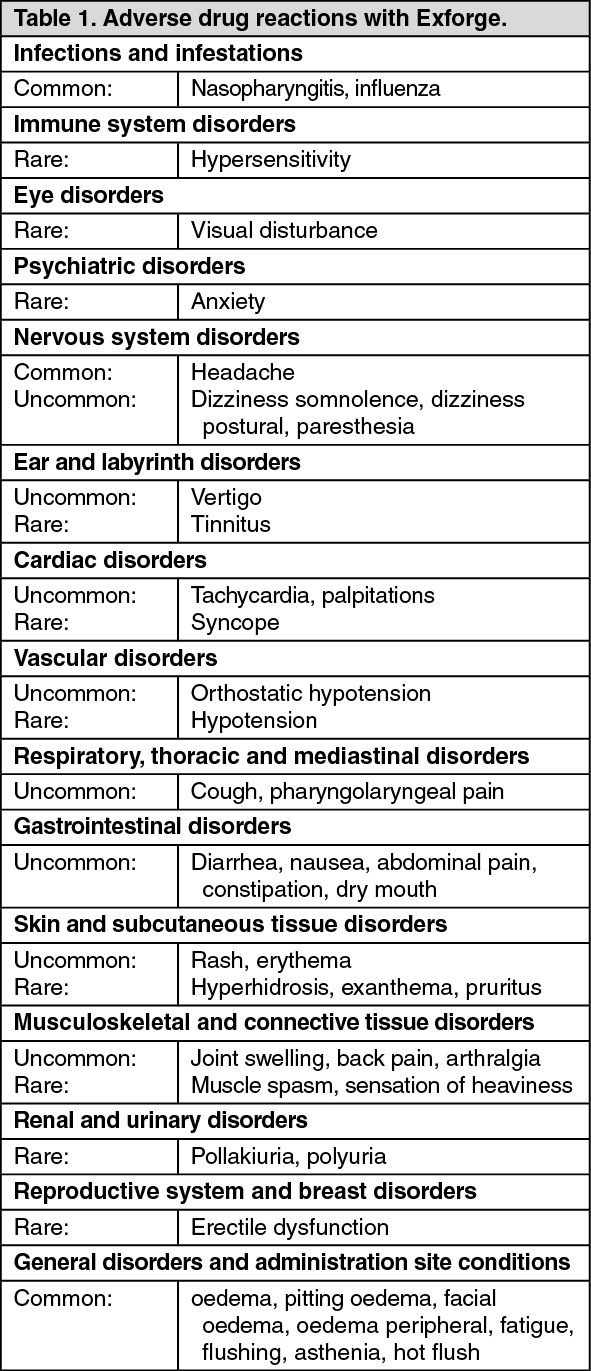
Adverse drug reactions or adverse experiences (Table 1 and Table 2) are ranked under heading of frequency, the most frequent first, using the following convention: very common (≥ 1/10); common (≥ 1/100, < 1/10); uncommon (≥ 1/1,000, < 1/100); rare (≥ 1/10,000, < 1/1,000) very rare (< 1/10,000), including isolated reports. Within each frequency grouping, adverse reactions are ranked in order of decreasing seriousness. (See Table 1.)
 Click on icon to see table/diagram/image
Additional information on the combination:
Click on icon to see table/diagram/image
Additional information on the combination: In double-blind, active- or placebo-controlled completed clinical trials, the incidence of peripheral edema was statistically lower in patients treated with the combination (5.8%) than in patients treated with amlodipine monotherapy (9%).
Laboratory evaluation: Very few hypertensive patients treated with valsartan/amlodipine showed notable changes in laboratory test results from baseline. There was a slightly higher incidence of notably increased blood urea nitrogen in the amlodipine/valsartan (5.5 %) and valsartan monotherapy (5.5%) groups as compared to the placebo group (4.5%).
Additional information on individual components: Adverse reactions previously reported with one of the individual components may occur with Exforge even if not observed in clinical trials.
Amlodipine: Other additional adverse experiences reported in clinical trials with amlodipine monotherapy, irrespective of their causal association with the study drug, are presented in Table 2: Because amlodipine clinical trials were conducted under widely varying conditions, adverse experience rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. (See Table 2.)
Click on icon to see table/diagram/image
Valsartan: Other ADRs reported from clinical studies, post-marketing experience and laboratory findings in hypertension indication are presented in Table 3 according to system organ class.
For all the ADRs reported from post-marketing experience and laboratory findings, it is not possible to apply any ADR frequency and therefore they are mentioned with a "not known" frequency. (See Table 3.)
Click on icon to see table/diagram/image
The following events have also been observed during clinical trials in hypertensive patients irrespective of their causal association with the study drug: Insomnia, libido decrease, pharyngitis, rhinitis, sinusitis, upper respiratory tract infection, viral infections.
Amlodipine: Simvastatin: Co-administration of multiple doses of 10 mg of amlodipine with 80 mg simvastatin resulted in a 77% increase in exposure to simvastatin compared to simvastatin alone. It is recommended to limit the dose of simvastatin to 20 mg daily in patients on amlodipine.
CYP3A4 Inhibitors: Co-administration of a 180 mg daily dose of diltiazem with 5 mg amlodipine in elderly hypertensive patients resulted in a 1.6 fold increase in amlodipine systemic exposure. However, strong inhibitors of CYP3A4 (e.g., ketoconazole, itraconazole, ritonavir) may increase the plasma concentrations of amlodipine to a greater extent than diltiazem. Caution should therefore be exercised when co-administering amlodipine with CYP3A4 inhibitors.
Grapefruit Juice: The exposure of amlodipine may be increased when co-administered with grapefruit juice due to CYP3A4 inhibition. However, co-administration of 240 mL of grapefruit juice with a single oral dose of amlodipine 10 mg in 20 healthy volunteers had no significant effect on the pharmacokinetics of amlodipine.
CYP3A4 Inducers: No information is available on the quantitative effects of CYP3A4 inducers on amlodipine. Patients should be monitored for adequate clinical effect when amlodipine is co-administered with CYP3A4 inducers (e.g. rifampicin, Hypericum perforatum).
In monotherapy, amlodipine has been safely administered with thiazide diuretics, beta-blockers, angiotensin-converting enzyme inhibitors, long-acting nitrates, sublingual nitroglycerin, digoxin, warfarin, atorvastatin, sildenafil, maalox (Aluminium hydroxide gel, Magnesium hydroxide and Simeticone), cimetidine, non-steroidal anti-inflammatory drugs, antibiotics, and oral hypoglycemic drugs.
Valsartan: Dual blockade of the renin-angiotensin system (RAS) with ARBs, ACEIs or aliskiren: The concomitant use of ARBs, including valsartan, with other agents acting on the RAS is associated with an increased incidence of hypotension, hyperkalaemia, and changes in renal function compared to monotherapy. It is recommended to monitor blood pressure, renal function and electrolytes in patients on Exforge and other agents that affect the RAS (see PRECAUTIONS).
The concomitant use of ARBs - including valsartan - or of ACEIs with aliskiren, should be avoided in patients with severe renal impairment (GFR < 30 mL/min) (see PRECAUTIONS).
The concomitant use of ARBs including valsartan, or ACEIs, with aliskiren is contraindicated in patients with Type 2 diabetes mellitus (see CONTRAINDICATIONS).
Potassium: Concomitant use with potassium supplements, potassium-sparing diuretics, salt substitutes containing potassium, or other drugs that may increase potassium levels (heparin, etc.) requires caution and frequent monitoring of potassium levels.
Non-Steroidal Anti-Inflammatory Agents (NSAIDs) including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors): When angiotensin II antagonists are administered simultaneously with NSAIDs, attenuation of the antihypertensive effect may occur. Furthermore, in patients who are elderly, volume-depleted (including those on diuretic therapy), or have compromised renal function, concomitant use of angiotensin II antagonists and NSAIDs may lead to an increased risk of worsening of renal function. Therefore, monitoring of renal function is recommended when initiating or modifying the treatment in patients on valsartan who are taking NSAIDs concomitantly.
Lithium: Reversible increases in serum lithium concentrations and toxicity have been reported during concomitant administration of lithium with ACE inhibitors or angiotensin II receptor antagonists including Exforge. Therefore, careful monitoring of serum lithium levels is recommended during concomitant use. If a diuretic is also used, the risk of lithium toxicity may presumably be increased further with Exforge.
Transporters: The results from an in vitro study with human liver tissue indicate that valsartan is a substrate of the hepatic uptake transporter OATP1B1 and the hepatic efflux transporter MRP2. Co-administration of inhibitors of the uptake transporter (e.g rifampin, ciclosporin) or efflux transporter (e.g. ritonavir) may increase the systemic exposure to valsartan.
In monotherapy with valsartan, no drug interactions of clinical significance have been found with the following drugs: cimetidine, warfarin, furosemide, digoxin, atenolol, indomethacin, hydrochlorothiazide, amlodipine, glibenclamide.
Incompatibilities: Not applicable.
Instructions for Use and Handling: No special requirements.
C09DB01 - valsartan and amlodipine ; Belongs to the class of angiotensin II receptor blockers (ARBs) and calcium channel blockers. Used in the treatment of cardiovascular disease.
Exforge 10/160 mg FC tab
14's;28's;7's
Exforge 5/160 mg FC tab
14's;28's;7's
Exforge 5/80 mg FC tab
14's;28's;7's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image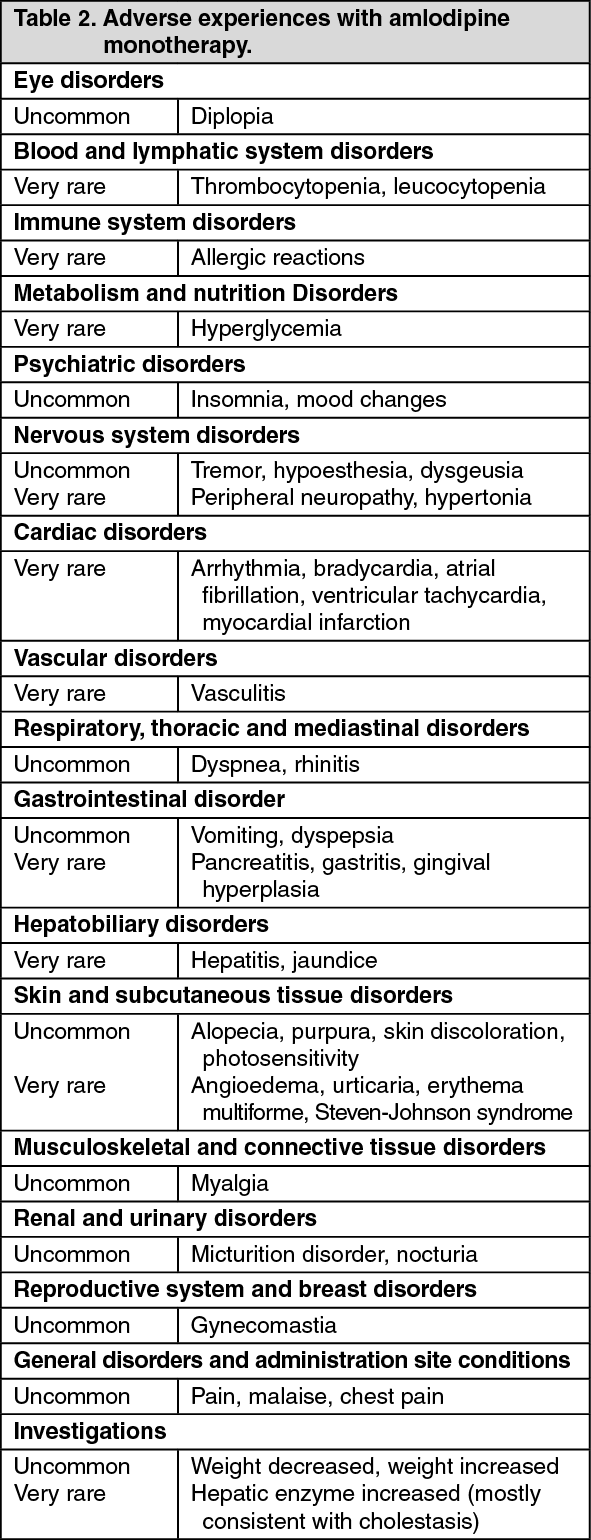 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image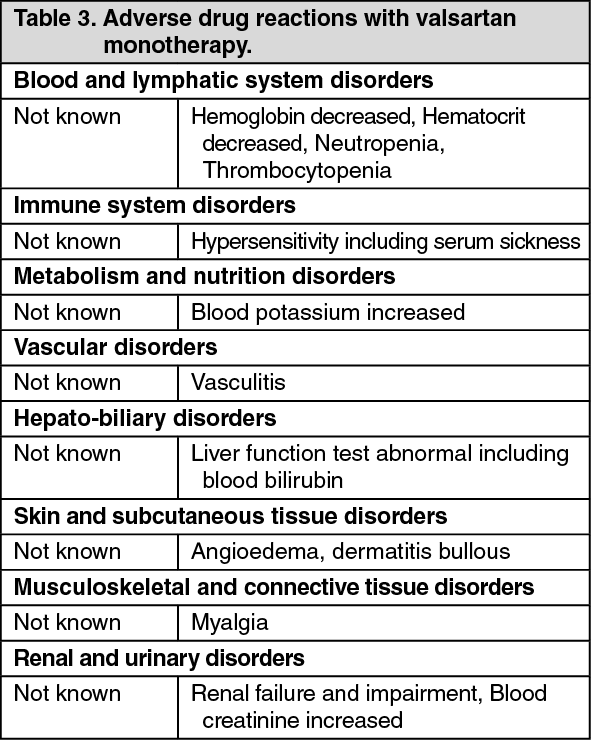 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
