


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Darifenacin is a selective muscarinic M3 receptor antagonist (M3 SRA) in vitro. The M3 receptor is the major subtype that controls urinary bladder muscle contraction. It is not known whether this selectivity for the M3 receptor translates into any clinical advantage when treating symptoms of overactive bladder syndrome.
Cystometric studies performed with darifenacin in patients with involuntary bladder contractions showed increased bladder capacity, increased volume threshold for unstable contractions and diminished frequency of unstable detrusor contractions.
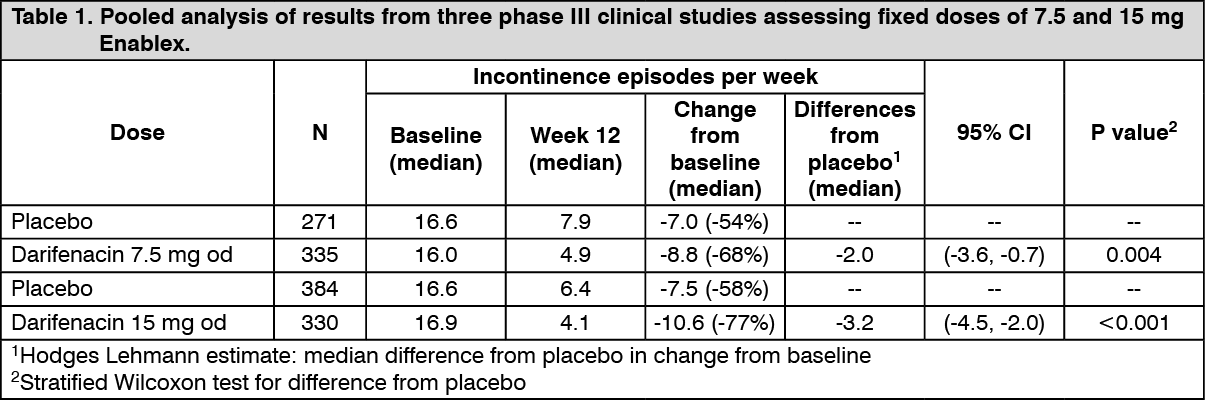
Treatment with Enablex administered at dosages of 7.5 mg and 15 mg daily has been investigated in four double-blind, phase III, randomised, controlled clinical studies in male and female patients with symptoms of overactive bladder. As seen in Table 1 as follows, a pooled analysis of 3 of the studies for the treatment with both Enablex 7.5 mg and 15 mg provided a statistically significant improvement in the primary endpoint, reduction in incontinence episodes, versus placebo (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEnablex 7.5 mg and 15 mg doses significantly reduced both the severity and number of urinary urgency episodes and the number of micturitions, while significantly increasing the mean volume voided from baseline.
Enablex 7.5 mg and 15 mg were associated with statistically significant improvements over placebo in some aspects of quality of life as measured by the Kings health questionnaire including incontinence impact, role limitations, social limitations and severity measures.
For both doses of 7.5 mg and 15 mg, the percentage median reduction from baseline in the number of incontinence episodes per week was similar between males and females. The observed differences from placebo for males in terms of percentage and absolute reductions in incontinence episodes was lower than for females.
The effect of treatment with 15 mg and 75 mg of darifenacin on QT/QTc interval was evaluated in a study in 179 healthy adults (44% male: 56% females) aged 18 to 65 for 6 days (to steady state). Therapeutic and supra-therapeutic doses of darifenacin resulted in no increase in QT/QTc interval prolongation from baseline compared to placebo at maximum darifenacin exposure.
Pharmacokinetics: Absorption: The mean oral bioavailability of darifenacin at steady state is estimated to be 15% and 19% for 7.5 and 15 mg tablets, respectively. Darifenacin is completely (>98%) absorbed after oral administration, although oral bioavailability is limited by first-pass metabolism (see Biotransformation/Metabolism as follows). Maximum plasma levels are reached approximately 7 hours after administration of the prolonged-release tablets and steady-state plasma levels are achieved by the sixth day of administration. At steady state, peak-to-trough fluctuations in darifenacin concentrations are small (peak to trough fluctuations: 0.87 for 7.5 mg and 0.76 for 15 mg), thereby maintaining therapeutic plasma levels over the dosing interval. Food had no effect on darifenacin pharmacokinetics during multiple-dose administration of prolonged-release tablets.
Distribution: Darifenacin is a lipophilic base and is 98% bound to plasma proteins (primarily to alpha-1-acid-glycoprotein). The steady-state volume of distribution (Vss) is estimated to be 163 litres.
Biotransformation/Metabolism: Darifenacin is extensively metabolised by the liver following oral administration.
Metabolism is mediated by cytochrome P450 enzymes CYP2D6 and CYP3A4. The three main metabolic routes are as follows: I. monohydroxylation in the dihydrobenzofuran ring; II. dihydrobenzofuran ring opening; II. N-dealkylation of the pyrrolidine nitrogen.
The initial products of the hydroxylation and N-dealkylation pathways are major circulating metabolites but none contributes significantly to the overall clinical effect of darifenacin.
Variability in metabolism: A subset of individuals (approximately 7% of the Caucasian population) is devoid of CYP2D6 enzyme activity. Therefore, the metabolism of darifenacin in these poor metabolisers will be principally mediated via CYP3A4. Individuals with full CYP2D6 activity are referred to as extensive metabolisers. The darifenacin ratios (poor metabolisers: extensive metabolisers) for Cmax and AUC following darifenacin 15 mg once-daily at steady state were 1.9 and 1.7, respectively.
Population pharmacokinetic analyses of Phase 3 data indicated that on average steady-state exposure is 66% higher in poor metabolisers than in extensive metabolisers. However, there is considerable overlap between the ranges of exposures seen in these two populations (see DOSAGE & ADMINISTRATION) and clinical experience confirms that there are no special dosing requirements for poor metabolisers.
Elimination: Following administration of an oral dose of 14C-darifenacin solution to healthy volunteers, approximately 60% of the radioactivity was recovered in the urine and 40% in the faeces. Only a small percentage of the excreted dose was unchanged darifenacin (3%). Estimated darifenacin clearance is 40 litres/hour for extensive metabolisers and 32 litres/hour for poor metabolisers. The elimination half-life of darifenacin following chronic dosing is approximately 13-19 hours.
Special Population: Gender: No special dosage requirements are necessary based on gender. A population pharmacokinetic analysis of patient data indicated that darifenacin exposure was 23% lower in males than females.
In clinical studies, the safety and efficacy profiles were not affected by gender.
Geriatric patients: There are no special dosage requirements for the elderly.
A population pharmacokinetic analysis of patient data indicated a trend for clearance to decrease with age (19% per decade based on Phase III population pharmacokinetic analysis of patients aged 60-89 years). The safety and efficacy profiles were not affected by age.
Paediatric patients: The pharmacokinetics of darifenacin have not been studied in the paediatric population.
Renal insufficiency: There are no special dosage requirements for patients with renal impairment. A small study of subjects (n=24) with varying degrees of renal impairment (creatinine clearance between 10 and 136 ml/min) given darifenacin 15 mg once daily to steady state demonstrated no relationship between renal function and darifenacin clearance.
Hepatic insufficiency: Darifenacin pharmacokinetics were investigated in subjects with mild (Child Pugh A) or moderate (Child Pugh B) impairment of hepatic function given darifenacin 15 mg once daily to steady state. Mild hepatic impairment had no effect on the pharmacokinetics of darifenacin. However, protein binding of darifenacin was affected by moderate hepatic impairment. After adjusting for plasma protein binding, unbound darifenacin exposure was estimated to be 4.7-fold higher in subjects with moderate hepatic impairment than subjects with normal hepatic function.
Toxicology: Preclinical safety data: Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and toxicity to reproduction.
Carcinogenicity studies with darifenacin were conducted in mice and rats. No evidence of drug related carcinogenicity was revealed in a 24-month study in mice at dietary doses up to 100 mg/kg/day or approximately 32 times the estimated human free AUC0-24h reached with 15 mg, the maximum recommended human dose (AUC at MRHD) and in a 24-month study in rats at doses up to 15 mg/kg/day or up to approximately 12 times the AUC at MRHD in female rats and approximately 8 times the AUC at MRHD in male rats.
Darifenacin was not mutagenic in the bacterial mutation assays (Ames test) and the Chinese hamster ovary assay, and not clastogenic in the human lymphocyte assay, and the in vivo mouse bone marrow cytogenetics assay.
There was no evidence for effects on fertility in male or female rats treated at oral doses up to 50 mg/kg/day. Exposures in this study correspond to approximately 78 times the AUC at MRHD.
Darifenacin was not teratogenic in rats and rabbits at doses up to 50 and 30 mg/kg/day respectively. At the dose of 50 mg/kg in rats, there was a delay in the ossification of the sacral and caudal vertebrae which was not observed at the lower doses of 3 and 10 mg/kg.
Exposure in this study at 50 mg/kg corresponds to approximately 59 times the AUC at MRHD. At the dose of 30 mg/kg in rabbits, darifenacin was shown to increase post-implantation loss but not at the lower doses tested (3 and 10 mg/kg). Exposure to unbound drug at 30 mg/kg in this study corresponds to approximately 28 times the AUC at MRHD.