Pharmacotherapeutic group: Immunosuppressants, Selective immunosuppressants.
ATC Code: L04AA31.
Pharmacology: Pharmacodynamics: Mechanism of action: Teriflunomide is an immunomodulatory agent with anti-inflammatory properties that selectively and reversibly inhibits the mitochondrial enzyme dihydroorotate dehydrogenase (DHO-DH), which functionally connects with the respiratory chain. As a consequence of the inhibition, teriflunomide generally reduces the proliferation of dividing cells that depend on de novo synthesis of pyrimidine to expand. The exact mechanism by which teriflunomide exerts its therapeutic effect in MS is not fully understood, but this is mediated by a reduced number of T-lymphocytes.
Pharmacodynamic effects: Immune system: Effects on immune cell numbers in the blood: In the placebo-controlled studies, teriflunomide 14 mg once a day led to a mild mean reduction in lymphocyte count, of less than 0.3 x 10
9/l, which occurred over the first 3 months of treatment and levels were maintained until the end of the treatment.
Potential to prolong the QT interval: In a placebo-controlled thorough QT study performed in healthy subjects, teriflunomide at mean steady-state concentrations did not show any potential for prolonging the QTcF interval compared with placebo: the largest time matched mean difference between teriflunomide and placebo was 3.45 ms with the upper bound of the 90% CI being 6.45 ms.
Effect on renal tubular functions: In the placebo-controlled studies, mean decreases in serum uric acid at a range of 20 to 30% were observed in patients treated with teriflunomide compared to placebo. Mean decrease in serum phosphorus was around 10% in the teriflunomide group compared to placebo. These effects are considered to be related to increase in renal tubular excretion and not related to changes in glomerular functions.
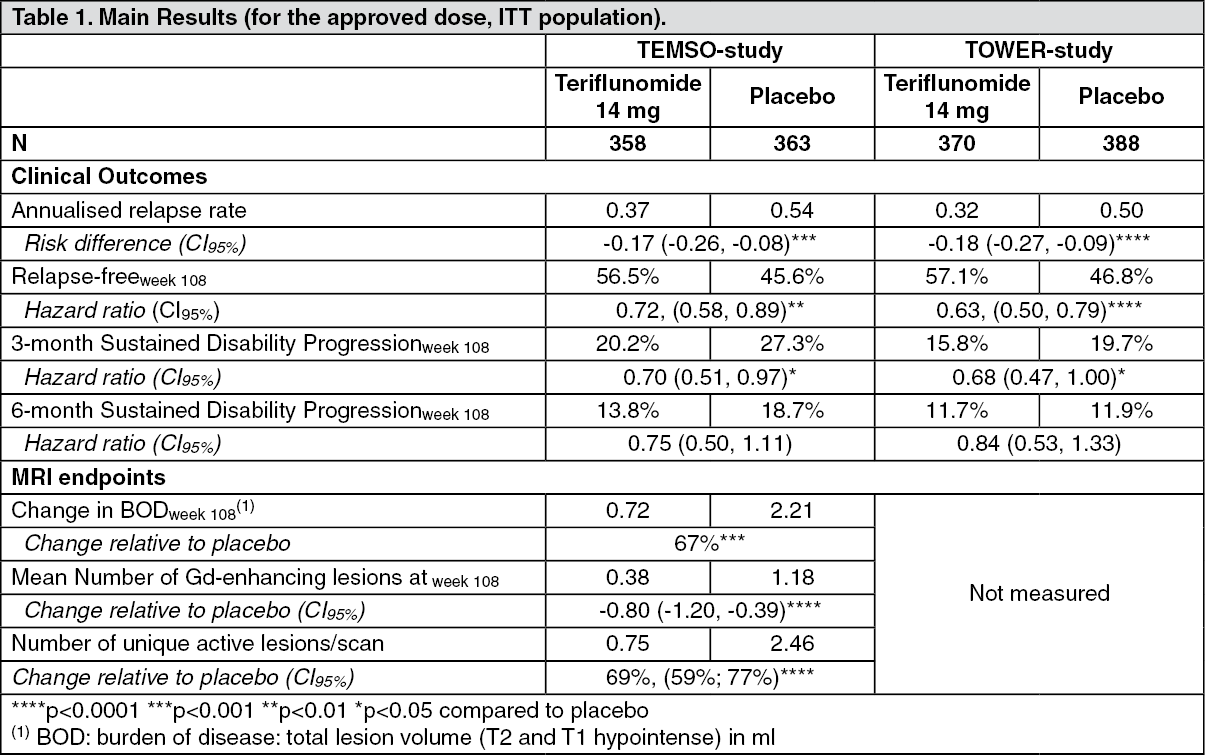
Clinical efficacy and safety: The efficacy of AUBAGIO was demonstrated in two placebo controlled studies, the TEMSO and the TOWER study, that evaluated once daily doses of teriflunomide 7 mg and 14 mg in patients with RMS.
A total of 1088 patients with RMS were randomised in TEMSO to receive 7 mg (n=366) or 14 mg (n=359) of teriflunomide or placebo (n=363) for 108 weeks duration. All patients had a definite diagnosis of MS (based on McDonald criteria (2001)), exhibited a relapsing clinical course, with or without progression, and experienced at least 1 relapse over the year preceding the trial or at least 2 relapses over the 2 years preceding the trial. At entry, patients had an Expanded Disability Status Scale (EDSS) score ≤5.5.
The mean age of the study population was 37.9 years. The majority of patients had relapsing-remitting multiple sclerosis (91.5%), but a subgroup of patients had secondary progressive (4.7%) or progressive relapsing multiple sclerosis (3.9%). The mean number of relapses within the year before study inclusion was 1.4 with 36.2% of patients having gadolinium-enhancing lesions at baseline. The median EDSS score at baseline was 2.50; 249 patients (22.9%) had an EDSS score > 3.5 at baseline. The mean duration of disease, since first symptoms, was 8.7 years. A majority of patients (73%) had not received disease-modifying therapy during the 2 years before study entry. The study results are shown in Table 1.
Long term follow-up results from TEMSO long term extension safety study (overall median treatment duration approximately 5 years, maximum treatment duration approximately 8.5 years) did not present any new or unexpected safety findings.
A total of 1169 patients with RMS were randomised in TOWER to receive 7 mg (n=408) or 14 mg (n=372) of teriflunomide or placebo (n=389) for a variable treatment duration ending at 48 weeks after last patient randomised. All patients had a definite diagnosis of MS (based on McDonald criteria (2005)), exhibited a relapsing clinical course, with or without progression, and experienced at least 1 relapse over the year preceding the trial or at least 2 relapses over the 2 years preceding the trial. At entry, patients had an Expanded Disability Status Scale (EDSS) score ≤5.5.
The mean age of the study population was 37.9 years. The majority of patients had relapsing-remitting multiple sclerosis (97.5%), but a subgroup of patients had secondary progressive (0.8%) or progressive relapsing multiple sclerosis (1.7%). The mean number of relapses within the year before study inclusion was 1.4. Gadolinium-enhancing lesions at baseline: no data. The median EDSS score at baseline was 2.50; 298 patients (25.5%) had an EDSS score > 3.5 at baseline. The mean duration of disease, since first symptoms, was 8.0 years. A majority of patients (67.2%) had not received disease-modifying therapy during the 2 years before study entry. The study results are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Efficacy in patients with high disease activity: A consistent treatment effect on relapses and time to 3-month sustained disability progression in a subgroup of patients in TEMSO (n=127) with high disease activity was observed. Due to the design of the study, high disease activity was defined as 2 or more relapses in one year, and with one or more Gd-enhancing lesion on brain MRI. No similar subgroup analysis was performed in TOWER as no MRI data were obtained. No data are available in patients who have failed to respond to a full and adequate course (normally at least one year of treatment) of beta-interferon, having had at least 1 relapse in the previous year while on therapy, and at least 9 T2-hyperintense lesions in cranial MRI or at least 1 Gd-enhancing lesion, or patients having an unchanged or increased relapse rate in the prior year as compared to the previous 2 years.
TOPIC was a double-blind, placebo-controlled study that evaluated once daily doses of teriflunomide 7 mg and 14 mg for up to 108 weeks in patients with first clinical demyelinating event (mean age 32.1 years). The primary endpoint was time to a second clinical episode (relapse). A total of 618 patients were randomized to receive 7 mg (n=205) or 14 mg (n=216) of teriflunomide or placebo (n=197). The risk of a second clinical attack over two years was 35.9% in the placebo group and 24.0% in the teriflunomide 14 mg treatment group (hazard ratio: 0.57, 95% confidence interval: 0.38 to 0.87, p=0.0087). The results from the TOPIC study confirmed the efficacy of teriflunomide in RRMS (including early RRMS with first clinical demyelinating event and MRI lesions disseminated in time and space).
Teriflunomide effectiveness was compared to that of a subcutaneous interferon beta-1a (at the recommended dose of 44 μg three times a week) in 324 randomised patients in a study (TENERE) with minimum treatment duration of 48 weeks (maximum 114 weeks). The risk of failure (confirmed relapse or permanent treatment discontinuation whichever came first) was the primary endpoint. The number of patients with permanent treatment discontinuation in the teriflunomide 14 mg group was 22 out of 111 (19.8%), the reasons being adverse events (10.8%), lack of efficacy (3.6%), other reason (4.5%) and lost to follow-up (0.9%). The number of patients with permanent treatment discontinuation in the subcutaneous interferon beta-1a group was 30 out of 104 (28.8%), the reasons being adverse events (21.2%), lack of efficacy (1.9%), other reason (4.8%) and poor compliance to protocol (1%). Teriflunomide 14 mg/day was not superior to interferon beta-1a on the primary endpoint: the estimated percentage of patients with treatment failure at 96 weeks using the Kaplan-Meier method was 41.1% versus 44.4% (teriflunomide 14 mg versus interferon beta-1a group, p=0.595).
Pharmacokinetics: Absorption: Median time to reach maximum plasma concentrations occurs between 1 to 4 hours post-dose following repeated oral administration of teriflunomide, with high bioavailability (approximately 100%).
Food does not have a clinically relevant effect on teriflunomide pharmacokinetics.
From the mean predicted pharmacokinetic parameters calculated from the population pharmacokinetic (PopPK) analysis using data from healthy volunteers and MS patients, there is a slow approach to steady-state concentration (i.e., approximately 100 days (3.5 months) to attain 95% of steady-state concentrations) and the estimated AUC accumulation ratio is approximately 34-fold.
Distribution: Teriflunomide is extensively bound to plasma protein (>99%), probably albumin and is mainly distributed in plasma. The volume of distribution is 11 l after a single intravenous (IV) administration. However, this is most likely an underestimation since extensive organ distribution was observed in rats.
Biotransformation: Teriflunomide is moderately metabolised and is the only component detected in plasma. The primary biotransformation pathway for teriflunomide is hydrolysis with oxidation being a minor pathway. Secondary pathways involve oxidation, N-acetylation and sulfate conjugation.
Elimination: Teriflunomide is excreted in the gastrointestinal tract mainly through the bile as unchanged medicinal product and most likely by direct secretion. Teriflunomide is a substrate of the efflux transporter BCRP, which could be involved in direct secretion. Over 21 days, 60.1% of the administered dose is excreted via feces (37.5%) and urine (22.6%). After the rapid elimination procedure with cholestyramine, an additional 23.1% was recovered (mostly in feces). Based on individual prediction of pharmacokinetic parameters using the PopPK model of teriflunomide in healthy volunteers and MS patients, median t
1/2z was approximately 19 days after repeated doses of 14 mg. After a single IV administration, the total body clearance of teriflunomide is 30.5 ml/h.
Accelerated Elimination Procedure: Cholestyramine and activated charcoal: The elimination of teriflunomide from the circulation can be accelerated by administration of cholestyramine or activated charcoal, presumably by interrupting the reabsorption processes at the intestinal level.
Teriflunomide concentrations measured during an 11-day procedure to accelerate teriflunomide elimination with either 8 g cholestyramine three times a day, 4 g cholestyramine three times a day or 50 g activated charcoal twice a day following cessation of teriflunomide treatment have shown that these regimens were effective in accelerating teriflunomide elimination, leading to more than 98% decrease in teriflunomide plasma concentrations, with cholestyramine being faster than charcoal. Following discontinuation of teriflunomide and the administration of cholestyramine 8 g three times a day, the plasma concentration of teriflunomide is reduced 52% at the end of day 1, 91% at the end of day 3, 99.2% at the end of day 7, and 99.9% at the completion of day 11. The choice between the 3 elimination procedures should depend on the patient's tolerability. If cholestyramine 8 g three times a day is not well-tolerated, cholestyramine 4 g three times a day can be used. Alternatively, activated charcoal may also be used (the 11 days do not need to be consecutive unless there is a need to lower teriflunomide plasma concentration rapidly).
Linearity/non-linearity: Systemic exposure increases in a dose proportional manner after oral administration teriflunomide from 7 to 14 mg.
Characteristics in specific groups of patients: Gender, Elderly, Paediatric patients: Several sources of intrinsic variability were identified in healthy subjects and MS patients based on the PopPK analysis: age, body weight, gender, race, and albumin and bilirubin levels. Nevertheless, their impact remains limited (≤ 31%).
Hepatic impairment: Mild and moderate hepatic impairment had no impact on the pharmacokinetic of teriflunomide. Therefore no dose adjustment is anticipated in mild and moderate hepatic-impaired patients. However, teriflunomide is a contraindicated in patients with severe hepatic impairment (see Dosage & Administration and Contraindications).
Renal impairment: Severe renal impairment had no impact on the pharmacokinetic of teriflunomide. Therefore no dose adjustment is anticipated in mild, moderate and severe renal-impaired patients.
Toxicology: Preclinical safety data: Repeated oral administration of teriflunomide to mice, rats and dogs for up to 3, 6, and 12 months, respectively, revealed that the major targets of toxicity were the bone marrow, lymphoid organs, oral cavity/gastro intestinal tract, reproductive organs, and pancreas. Evidence of an oxidative effect on red blood cells was also observed. Anemia, decreased platelet counts and effects on the immune system, including leukopenia, lymphopenia and secondary infections, were related to the effects on the bone marrow and/or lymphoid organs. The majority of effects reflect the basic mode of action of the compound (inhibition of dividing cells). Animals are more sensitive to the pharmacology, and therefore toxicity, of teriflunomide than humans. As a result, toxicity in animals was found at exposures equivalent or below human therapeutic levels.
Teriflunomide was not mutagenic
in vitro or clastogenic
in vivo. Clastogenicity observed
in vitro was considered to be an indirect effect related to nucleotide pool imbalance resulting from the pharmacology of DHO-DH inhibition. The minor metabolite TFMA (4-trifluoromethylaniline) caused mutagenicity and clastogenicity
in vitro but not
in vivo.
No evidence of carcinogenicity was observed in rats and mice.
Fertility was unaffected in rats despite adverse effects of teriflunomide on male reproductive organs, including reduced sperm count. There were no external malformations in the offspring of male rats administered teriflunomide prior to mating with untreated female rats. Teriflunomide was embryotoxic and teratogenic in rats and rabbits at doses in the human therapeutic range. Adverse effects on the offspring were also seen when teriflunomide was administered to pregnant rats during gestation and lactation. The risk of male-mediated embryo-fetal toxicity through teriflunomide treatment is considered low. The estimated female plasma exposure via the semen of a treated patient is expected to be 100 times lower than the plasma exposure after 14 mg of oral teriflunomide.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
