


 Sign Out
Sign Out

The serum adalimumab concentration-efficacy relationship as measured by the American College of Rheumatology response criteria (ACR 20) appears to follow the Hill Emax equation as shown as follows: (see Figure 1.)
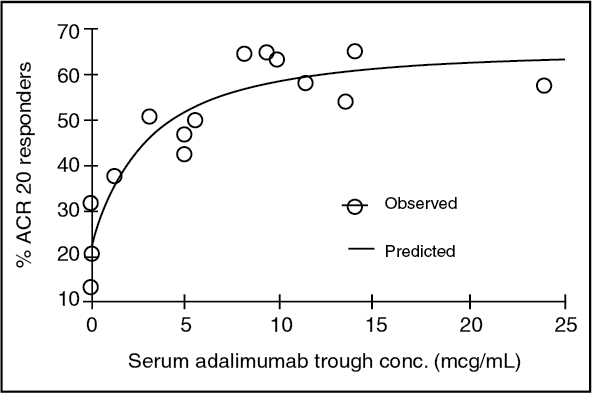 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEC50 estimates ranging from 0.8 to 1.4 mcg/mL were obtained through pharmacokinetic/pharmacodynamic modeling of swollen joint count, tender joint count and ACR 20 response from patients participating in Phase II and III trials.
General: Adalimumab binds specifically to TNF and neutralizes the biological function of TNF by blocking its interaction with the p55 and p75 cell surface TNF receptors. TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Elevated levels of TNF are found in the synovial fluid of rheumatoid arthritis, including juvenile idiopathic arthritis, psoriatic arthritis and ankylosing spondylitis patients and play an important role in both the pathologic inflammation and the joint destruction that are hallmarks of these diseases. Increased levels of TNF are also found in psoriasis (Ps) plaques. In plaque psoriasis, treatment with Adalimumab may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which Adalimumab exerts its clinical effects is unknown.
Adalimumab also modulates biological responses that are induced or regulated by TNF, including changes in the levels of adhesion molecules responsible for leukocyte migration (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 1-2 X 10-10M).
Clinical Trials: COMPARABILITY OF AMGEVITA WITH HUMIRA: CLINICAL TRIAL FOR RHEUMATOID ARTHRITIS (Study 20120262): The efficacy and safety of AMGEVITA compared with HUMIRA were assessed in a randomised active-control, double-blind study in patients ≥ 18 years of age with moderate to severe active rheumatoid arthritis with inadequate response to methotrexate. The patients had either rheumatoid factor or anti-cyclic citrullinated peptide positivity. The study evaluated 526 patients on stable doses of Methotrexate. Patients were randomised to receive 40 mg of AMGEVITA or HUMIRA subcutaneously every other week for up to 22 weeks.
The percent of AMGEVITA-treated subjects achieving ACR 20 at week 24 in the RA Study is shown in Table 1. At week 24, 74.6% (194/260) subjects in the AMGEVITA group and 72.4% (189/261) subjects in the HUMIRA group met the ACR 20 response criteria. The risk ratio (RR) of ACR 20 for AMGEVITA versus HUMIRA was 1.039 with the 2-sided 90% confidence interval (CI) of (0.954, 1.133). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe RR of ACR 20 primary endpoint was within the pre-specified margin and showed clinical equivalence between AMGEVITA and HUMIRA.
The results of the components of the ACR response criteria for RA ABP-Study 1 are shown in Table 2. ACR response rates and improvement in all components of ACR response showed an absence of clinically meaningful differences between the two groups at week 24. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe time course of ACR20 response is shown in Figure 2. (See Figure 2.)
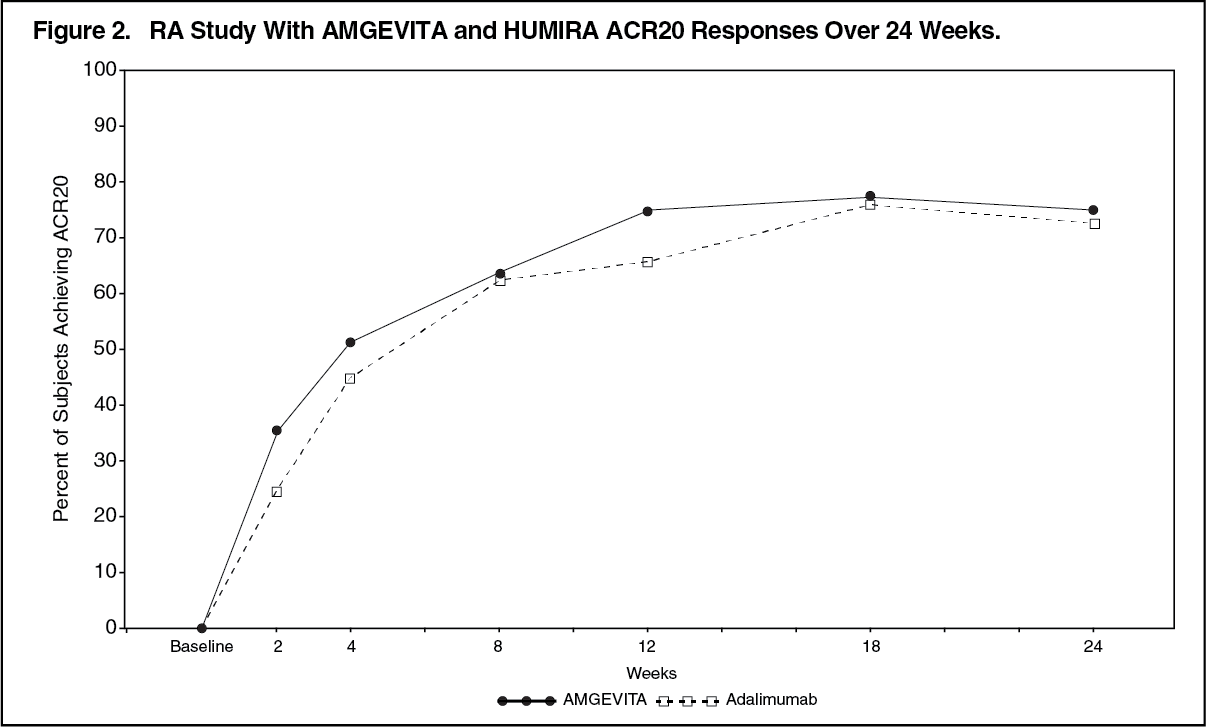 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCLINICAL TRIAL FOR PSORIASIS (Study 20120263): The efficacy and safety of AMGEVITA were assessed in a randomised active-control, double-blind study in 350 patients ≥ 18 years of age with moderate to severe plaque psoriasis (Ps) who were candidates for systemic therapy or phototherapy. Patients had stable moderate to severe plaque Ps for at least 6 months, a body surface area (BSA) ≥ 10%, and Psoriasis Area and Severity Index (PASI) ≥ 12 at study entry. The patients received AMGEVITA or HUMIRA at an initial loading dose of 80 mg administered SC on week 1/day1, followed by 40 mg SC given every other week starting one week after the loading dose. The PASI percent improvement from baseline was measured and compared with adalimumab (see Table 3) and it was within the pre-specified equivalence margin to demonstrate clinical equivalence between AMGEVITA and HUMIRA. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary endpoint was PASI percent improvement from baseline to week 16. At week 16, the PASI percent improvement from baseline was 80.9 in the AMGEVITA group and 83.1 in the HUMIRA group. The least-squares (LS) mean difference of PASI percent improvement from baseline to week 16 between AMGEVITA and HUMIRA was -2.18 with the 2-sided 95% CI of (-7.39, 3.02). The 95% CI was within the predefined equivalence margin, thus demonstrating clinical equivalence of AMGEVITA and HUMIRA.
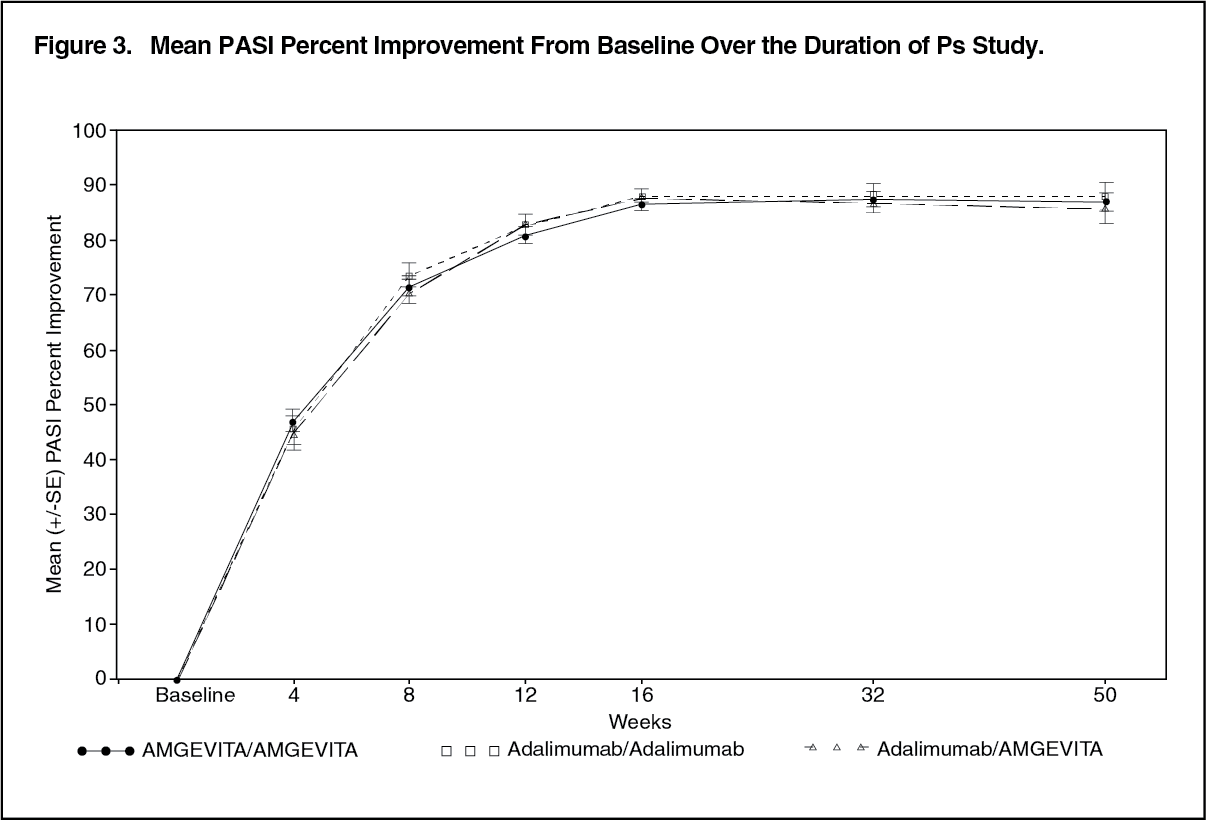
The Ps study was also designed to evaluate clinically meaningful differences in safety and immunogenicity in subjects who underwent a single transition from HUMIRA to AMGEVITA at week 16 and to provide a descriptive comparison with patients who continued on HUMIRA. The 350 subjects in the Ps study were initially randomised (1:1) to Treatment Group A (AMGEVITA) or Treatment Group B (HUMIRA). At week 16, subjects with a PASI 50 response (50% or better improvement) continued on study for up to 52 weeks. Subjects who continued treatment beyond week 16 were re-randomised in a blinded fashion such that all subjects initially randomised to Treatment Group A (AMGEVITA) continued treatment with AMGEVITA (AMGEVITA/AMGEVITA) and subjects initially randomised to Treatment Group B (HUMIRA) were re-randomised (1:1) to either continue treatment with HUMIRA, Treatment Group B1 (HUMIRA/HUMIRA) or were transitioned to AMGEVITA, Treatment Group B2 (HUMIRA/AMGEVITA). Subjects continued with their assigned treatment until week 48, when the last dose of assigned investigational product was administered and week 52 was the end of study.
The overall safety profile of the subjects who transitioned from HUMIRA to AMGEVITA was similar to the subjects who remained on HUMIRA throughout the study.
The mean PASI percent improvement from baseline over the duration of the study is shown in Figure 3. (See Figure 3.)
 Click on icon to see table/diagram/image
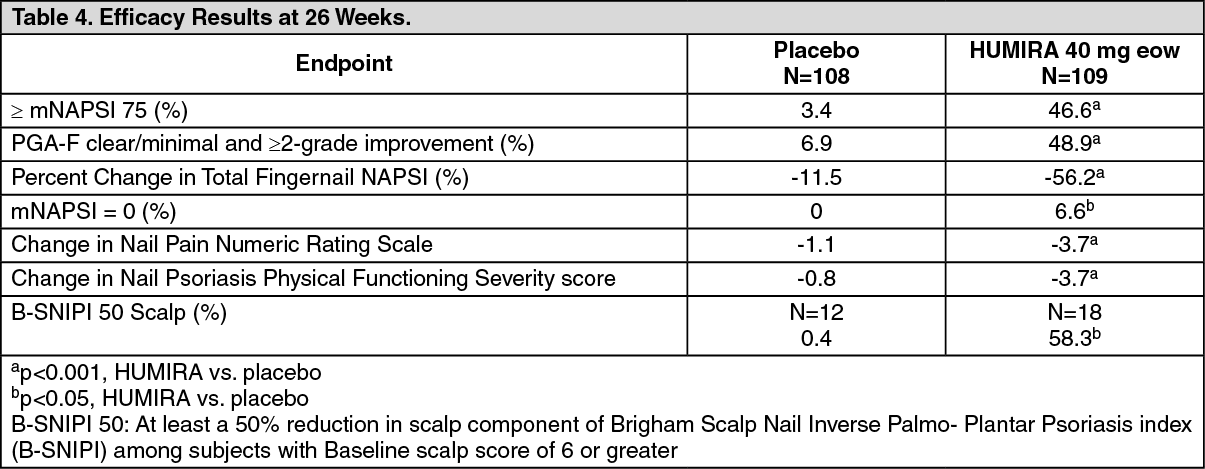
Click on icon to see table/diagram/imageCLINICAL STUDY ON NAIL PSORIASIS: Psoriasis Study IV compared the efficacy and safety of adalimumab versus placebo in 217 adult patients with moderate to severe nail psoriasis. Patients received an initial dose of 80 mg HUMIRA followed by 40 mg every other week (starting one week after the initial dose) or placebo for 26 weeks followed by open-label HUMIRA treatment for an additional 26 weeks. Nail psoriasis was assessed using the Modified Nail Psoriasis Severity Index (mNAPSI) and the Physician's Global Assessment of Fingernail Psoriasis (PGA-F). A statistically significantly higher proportion of patients randomized to HUMIRA achieved at least a 75% improvement in mNAPSI (mNAPSI 75) at Week 26, as compared with patients randomized to placebo (see Table 4). The percent improvement in NAPSI was statistically significantly greater in HUMIRA patients compared with placebo at Week 16 (44.2% vs 7.8%) and at Week 26 (56.2% vs 11.5%).
A statistically significant higher proportion of patients in the HUMIRA group achieved a PGA-F of "clear" or "minimal" with at least a 2-grade improvement from Baseline at Week 26 compared with placebo. In this study, HUMIRA demonstrated a treatment benefit in nail psoriasis patients with different extents of skin involvement (BSA≥10% and BSA<10% and ≥5%) and a statistically significant improvement in scalp psoriasis compared with placebo. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOf those who continued to receive adalimumab treatment until Week 52, 65.0% achieved mNAPSI 75 response and 61.3% achieved PGA-F response.
Adalimumab treated patients showed statistically significant improvements at Week 26 from baseline compared with placebo in the DLQI (Dermatology Life Quality Index). The mean decrease (improvement) from baseline at Week 26 was 8.0 in the HUMIRA group (N=94) and 1.9 in the placebo group (N=93).
IMMUNOGENICITY OF Adalimumab: Patients in rheumatoid arthritis studies I, II, and III were tested at multiple time points for anti-adalimumab antibodies during the 6 to 12 month period. Approximately 5.5% (58 of 1,062) of adult rheumatoid arthritis patients receiving adalimumab developed low-titre antibodies to adalimumab at least once during treatment, which were neutralising in vitro. Patients treated with concomitant MTX had a lower rate of antibody development than patients on adalimumab monotherapy (1% versus 12%). No apparent correlation of antibody development to adverse events was observed. With monotherapy, patients receiving fortnightly dosing may develop antibodies more frequently than those receiving weekly dosing. In patients receiving the recommended dosage of 40 mg fortnightly as monotherapy, the ACR 20 response was lower among antibody-positive patients than among antibody-negative patients. The long-term immunogenicity of adalimumab is unknown.
In pJIA Study I a greater percentage of patients developed antibodies to adalimumab compared to adult rheumatoid arthritis patients. Antibody formation was lower when adalimumab was given together with methotrexate in comparison with use as monotherapy. There was no apparent correlation between the presence of antibodies and adverse events. Anti-adalimumab antibodies were identified in 15.8% (27/171) of patients treated with adalimumab. In patients not given concomitant methotrexate, the incidence was 25.6% (22/86), compared to 5.9% (5/85) when adalimumab was used as add-on to methotrexate.
In pJIA Study II anti-adalimumab antibodies were identified in 7% (1/15) of patients, and the one patient was receiving concomitant methotrexate.
In patients with enthesitis-related arthritis, anti-adalimumab antibodies were identified in 11% (5/46) of patients treated with adalimumab. In patients not given concomitant methotrexate, the incidence was 14% (3/22), compared to 8% (2/24) when adalimumab was used as add-on to methotrexate.
In paediatric patients with moderately to severely active Crohn's disease, the rate of antibody development in patients receiving adalimumab was 3.3%.
In patients with ankylosing spondylitis, the rate of development of anti-adalimumab antibodies in adalimumab-treated patients was comparable to patients with rheumatoid arthritis. In patients with psoriatic arthritis, the rate of antibody development in patients receiving adalimumab monotherapy was comparable to patients with rheumatoid arthritis; however, in patients receiving concomitant methotrexate the rate was 7% compared to 1% in rheumatoid arthritis. The immunogenicity rate was 8% for psoriasis patients who were treated with adalimumab monotherapy.
In patients with Crohn's disease, anti-adalimumab antibodies were identified in 2.6% (7/269) of patients treated with adalimumab.
In patients with ulcerative colitis, anti-adalimumab antibodies were identified in 3.9% (19/487) of patients treated with adalimumab. However, due to the limitation of the assay conditions, antibodies to adalimumab could be detected only when serum adalimumab levels were <2 micrograms/mL. Among the patients whose serum adalimumab levels were <2 micrograms/mL (approximately 25% of total patients studied), the immunogenicity rate was 20.7%.
In plaque psoriasis patients on long-term adalimumab without concomitant methotrexate who participated in a withdrawal and retreatment study, the rate of anti-adalimumab antibodies after retreatment was similar to the rate observed prior to withdrawal.
In patients with paediatric psoriasis, anti-adalimumab antibodies were identified in 13% (5/38) of subjects treated with 0.8 mg/kg adalimumab monotherapy. 37 of the 38 subjects completed the initial double blind period (16 weeks) of Study M04-717, and one subject entered the long-term follow up period after Week 4.
In patients with moderate to severe hidradenitis suppurativa, anti-adalimumab antibodies were identified in 10/99 subjects (10.1%) treated with adalimumab.
In patients with non-infectious uveitis, anti-adalimumab antibodies were identified in 4.8% (12/249) of patients treated with adalimumab.
The data reflect the percentage of patients whose test results were considered positive for antibodies to adalimumab in an ELISA assay, and are highly dependent on the sensitivity and specificity of the assay. For these reasons, comparison of the incidence of antibodies to adalimumab with the incidence of antibodies to other products may be misleading.
IMMUNOGENICITY of AMGEVITA: Differences in assay methodology for measuring immunogenicity prevents direct comparison of immunogenicity rates between AMGEVITA and HUMIRA or other biologics in different studies. In the RA and Ps studies, binding ADA activity was determined using a bridging immunoassay and the neutralising ADA activity was determined using a TNFα-ligand binding based bioassay.
Immunogenicity in the RA study: Patients were tested at multiple time points for antibodies to AMGEVITA and HUMIRA during the 26-week study period. The incidence of developing binding antibodies was 38.3% (101/264) in the AMGEVITA group and 38.2% (100/262) in the HUMIRA group; the incidence of developing neutralising antibodies was 9.1% (24/264) in the AMGEVITA group and 11.1% (29/262) in the HUMIRA group. The immunogenicity profile of AMGEVITA was similar to HUMIRA.
Immunogenicity in the Ps study: Patients in the Ps study were tested at multiple time points for antibodies to HUMIRA and AMGEVITA during the 52-week study period. The incidence of developing binding antibodies through the duration of the study was 68.4% (104/152) in the AMGEVITA/AMGEVITA group, 74.7% (59/79) in the HUMIRA/HUMIRA group, and 72.7% (56/77) in the HUMIRA/AMGEVITA group; the incidence of developing neutralising antibodies was 13.8% (21/152) in the AMGEVITA/AMGEVITA group, 20.3% (16/79) in the HUMIRA/HUMIRA group, and 24.7% (19/77) in the HUMIRA/AMGEVITA group. The HUMIRA/AMGEVITA group reflects data for subjects exposed to both HUMIRA and AMGEVITA before and after the transition. The safety and immunogenicity profiles of patients who transitioned from HUMIRA to AMGEVITA were comparable to those who continued on HUMIRA until the end of the study (week 52).
Pharmacokinetics: Absorption: Following a single 40 mg subcutaneous (SC) administration of adalimumab to 59 healthy adult subjects, absorption and distribution of adalimumab was slow, with mean peak serum concentration being reached about five days after administration. The average absolute bioavailability of adalimumab estimated from three studies following a single 40 mg subcutaneous dose was 64%.
Distribution and Elimination: The single dose pharmacokinetics of adalimumab were determined in several studies with intravenous doses ranging from 0.25 to 10 mg/kg. The distribution volume (Vss) ranged from 4.7 to 6.0 L, indicating that adalimumab distributes approximately equally between the vascular and extravascular fluids. Adalimumab is slowly eliminated, with clearances typically under 12 mL/h. The mean terminal phase half-life was approximately two weeks, ranging from 10 to 20 days across studies. The clearance and half-life were relatively unchanged over the studied dose range, and the terminal half-life was similar after IV and SC administration. Adalimumab concentrations in the synovial fluid from several RA patients ranged from 31 to 96% of those in serum.
Steady-state pharmacokinetics: Accumulation of adalimumab was predictable based on the half-life following SC administration of 40 mg of adalimumab every other week to patients with RA, with mean steady-state trough concentrations of approximately 5 mcg/mL (without concomitant methotrexate (MTX)) and 8 to 9 mcg/mL (with concomitant MTX), respectively. The serum adalimumab trough levels at steady-state increased approximately proportionally with dose following 20, 40 and 80 mg every other week and every week SC dosing. In long-term studies with dosing more than two years, there was no evidence of changes in clearance over time.
In patients with psoriasis, the mean steady-state trough concentration was 5 mcg/mL during adalimumab 40 mg eow without concomitant methotrexate treatment.
In patients with hidradenitis suppurativa, a dose of 160 mg adalimumab on Week 0 followed by 80 mg on Week 2 achieved serum adalimumab trough concentrations of approximately 7 to 8 mcg/mL at Week 2 and Week 4. The mean steady-state trough concentration at Week 12 through Week 36 were approximately 8 to 10 mcg/mL during adalimumab 40 mg every week treatment.
In patients with uveitis, a loading dose of 80 mg adalimumab on Week 0 followed by 40 mg adalimumab every other week starting at Week 1, result in mean steady-state concentrations of approximately 8 to 10 mcg/mL.
Population pharmacokinetic analyses with data from over 1200 patients revealed that co-administration of MTX had an intrinsic effect on adalimumab apparent clearance (CL/F) (see Interactions). As expected, there was a trend towards higher apparent clearance of adalimumab with increasing body weight and in the presence of anti-adalimumab antibodies.
Other more minor factors were also identified; higher apparent clearance was predicted in patients receiving doses lower than the recommended dose, and in patients with high rheumatoid factor or CRP concentrations. These factors are not likely to be clinically important.
In patients with Crohn's disease, the loading dose of 160 mg adalimumab on Week 0 followed by 80 mg adalimumab on Week 2 achieves mean serum adalimumab trough levels of approximately 12 mcg/mL at Week 2 and Week 4. Mean steady-state trough levels of approximately 7 mcg/mL were observed at Week 24 and Week 56 in Crohn's disease patients after receiving a maintenance dose of 40 mg adalimumab every other week.
In patients with ulcerative colitis, a loading dose of 160 mg adalimumab on Week 0 followed by 80 mg adalimumab on Week 2 achieves serum adalimumab trough concentrations of approximately 12 mcg/mL during the induction period. Mean steady-state trough levels of approximately 8 mcg/mL were observed in ulcerative colitis patients who received a maintenance dose of 40 mg adalimumab every other week.
Special Populations: Pharmacokinetics in special populations were investigated using population pharmacokinetic analyses.
Geriatrics: Age appeared to have a minimal effect on adalimumab apparent clearance. From the population analyses, the mean weight-adjusted clearances in patients 40 to 65 years (n=850) and ≥65 years (n=287) were 0.33 and 0.30 mL/h/kg, respectively.
Pediatrics: Following the administration of 24 mg/m2 (up to a maximum of 40 mg) subcutaneously every other week to patients with polyarticular juvenile idiopathic arthritis (JIA) who were 4 to 17 years, the mean trough steady-state (values measured from Week 20 to 48) serum adalimumab concentration was 5.6 ± 5.6 μg/mL (102% CV) for adalimumab without concomitant methotrexate and 10.9 ± 5.2 μg/mL (47.7% CV) with concomitant methotrexate. The mean steady-state trough serum adalimumab concentrations for patients weighing <30 kg receiving 20 mg adalimumab subcutaneously every other week without concomitant methotrexate or with concomitant methotrexate were 6.8 μg/mL and 10.9 μg/mL, respectively. The mean steady-state trough serum adalimumab concentrations for patients weighing ≥30 kg receiving 40 mg adalimumab subcutaneously every other week without concomitant methotrexate or with concomitant methotrexate were 6.6 μg/mL and 8.1 μg/mL, respectively. In patients with polyarticular juvenile idiopathic arthritis (JIA) who were 2 to <4 years old or aged 4 and above weighing <15 kg dosed with adalimumab 24 mg/m2, the mean trough steady-state serum adalimumab concentrations was 6.0 ± 6.1 μg/ ml (101% CV) for adalimumab without concomitant methotrexate and 7.9 ± 5.6 μg/ml (71.2% CV) with concomitant methotrexate.
Following the administration of 24 mg/m2 (up to a maximum of 40 mg) subcutaneously every other week to patients with enthesitis-related arthritis, the mean trough steady-state (values measured at Week 24) serum adalimumab concentrations were 8.8 ± 6.6 μg/mL for adalimumab without concomitant methotrexate and 11.8 ± 4.3 μg/mL with concomitant methotrexate.
In pediatric patients with moderately to severely active Crohn's disease, the open-label adalimumab induction dose was 160/80 mg or 80/40 mg at Weeks 0 and 2, respectively, dependent on a body weight cut-off of 40 kg. At Week 4, patients were randomized 1:1 to either the Standard Dose (40/20 mg eow) or Low Dose (20/10 mg eow) maintenance treatment groups based on their body weight. The mean (±SD) serum adalimumab trough concentrations achieved at Week 4 were 15.7±6.6 μg/mL for patients ≥40 kg (160/80 mg) and 10.6±6.1 μg/mL for patients <40 kg (80/40 mg).
For patients who stayed on their randomized therapy, the mean (±SD) adalimumab trough concentrations at Week 52 were 9.5±5.6 μg/mL for the Standard Dose group and 3.5±2.2 μg/mL for the Low Dose group. The mean trough concentrations were maintained in patients who continued to receive adalimumab treatment eow for 52 weeks. For patients who dose escalated from eow to weekly regimen, the mean (±SD) serum concentrations of adalimumab at Week 52 were 15.3±11.4 μg/mL (40/20 mg, weekly) and 6.7±3.5 μg/mL (20/10 mg, weekly).
Following the administration of 0.8 mg/kg (up to a maximum of 40 mg) subcutaneously every other week to paediatric patients with chronic plaque psoriasis, the mean ± SD steady-state adalimumab trough concentration was approximately 7.4 ± 5.8 μg/mL (79% CV).
Gender: No gender-related pharmacokinetic differences were observed after correction for a patient's body weight.
Race: No differences in immunoglobulin clearance would be expected among races. From limited data in non-Caucasians, no important kinetic differences were observed for adalimumab.
Hepatic and Renal Insufficiency: No pharmacokinetic data are available in patients with hepatic or renal impairment.
Disease States: Healthy volunteers and patients with RA displayed similar adalimumab pharmacokinetics.
Comparability of AMGEVITA with HUMIRA: AMGEVITA is pharmacokinetically similar to HUMIRA.
Pharmacokinetic similarity was demonstrated between AMGEVITA and HUMIRA following administration of a single 40 mg dose subcutaneously in 203 healthy adult subjects. Pharmacokinetic parameters such as maximum serum concentrations and area under the serum concentration time curves were compared. According to the bioequivalence testing, the 90% confidence intervals of the geometric mean test-to-reference ratios for these parameters fell within the protocol-specified criteria of 0.8 to 1.25 and concluded pharmacokinetic similarity between AMGEVITA and HUMIRA.
Toxicology: Preclinical Safety Data: Preclinical data reveal no special hazard for humans based on studies of single dose toxicity, repeated dose toxicity, and genotoxicity.
Carcinogenesis, Mutagenesis, and Impairment of Fertility: Long-term animal studies of adalimumab have not been conducted to evaluate the carcinogenic potential or its effect on fertility.
No clastogenic or mutagenic effects of adalimumab were observed in the in vivo mouse micronucleus test or the Salmonella-Escherichia coli (Ames) assay, respectively.