Serious and or clinically significant adverse reactions described elsewhere in labeling include: Anaphylaxis and Hypersensitivity Reactions (see Precautions), Acute Respiratory Complications Associated with Administration (see Precautions), Risk of Acute Cardiorespiratory Failure (see Precautions), Infusion Reactions (see Precautions).
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most serious adverse reactions reported with ALDURAZYME treatment during clinical trials were anaphylactic and hypersensitivity reactions. Most adverse reactions reported in clinical trials were considered disease-related and unrelated to study drug. The most common adverse reactions were infusion reactions. The frequency of infusion reactions decreased over time with continued use of ALDURAZYME, and the majority of reactions were classified as being mild to moderate in severity. Most infusion reactions requiring intervention were ameliorated with slowing of the infusion rate, temporarily stopping the infusion, with or without administering additional treatments including antihistamines, antipyretics or both.
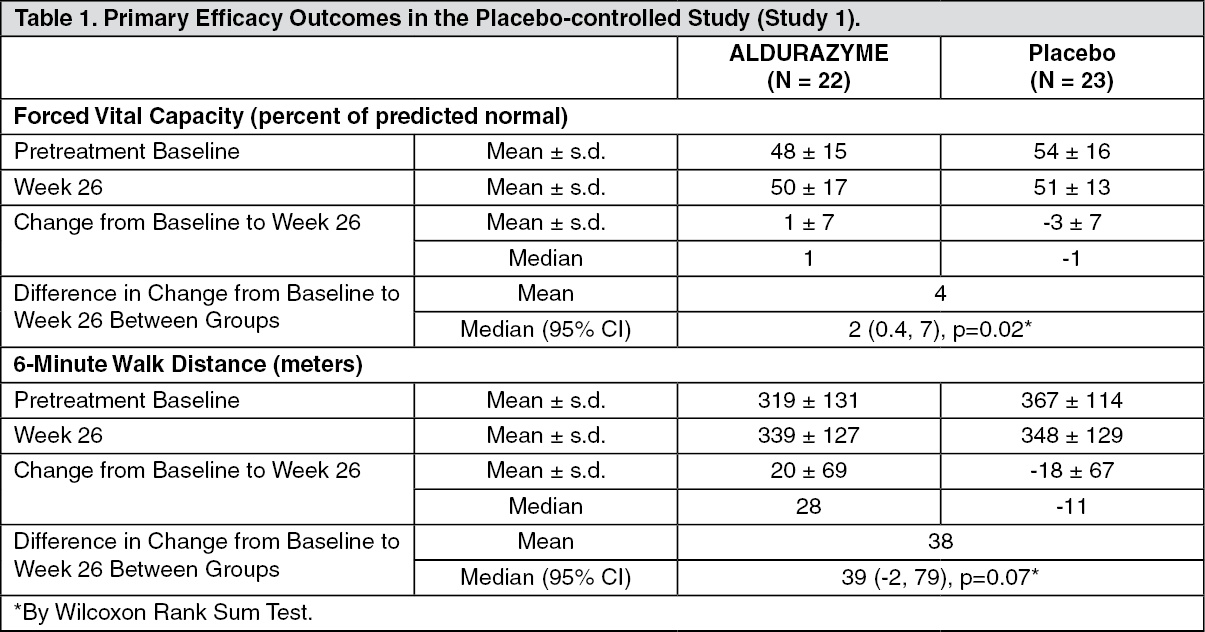
Clinical Trials in Patients 6 Years and Older: A 26-week, double-blind, placebo-controlled clinical study (Study 1) of ALDURAZYME was conducted in 45 patients with MPS I, ages 6 to 43 years old, gender evenly distributed (N = 23 females and 22 males). Of these 45 patients, 1 was clinically assessed as having Hurler form, 37 Hurler-Scheie, and 7 Scheie. Patients were randomized to receive either 0.58 mg/kg intravenously of ALDURAZYME per week for 26 weeks or placebo. All patients were treated with antipyretics and antihistamines prior to the infusions. Infusion reactions were reported in 32% (7 of 22) of ALDURAZYME treated patients. The most commonly reported infusion reactions regardless of treatment group were flushing, pyrexia, headache, and rash. Flushing occurred in 5 patients (23%) receiving ALDURAZYME; the other reactions were less frequent. Less common infusion reactions included angioedema (including face oedema), hypotension, paresthesia, feeling hot, hyperhidrosis, tachycardia, vomiting, back pain, and cough. Other reported adverse reactions included bronchospasm, dyspnea, urticaria and pruritus.
Table 4 enumerates adverse reactions and selected laboratory abnormalities that occurred during the placebo-controlled study (Study 1) that were reported in at least 2 patients more in the ALDURAZYME group than in the placebo group. (See Table 4).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
All 45 patients who completed the placebo-controlled study (Study 1) continued treatment in an open-label, uncontrolled extension study (Study 2). All patients received ALDURAZYME 0.58 mg/kg of body weight once weekly for up to 182 weeks. The most serious adverse reactions reported with ALDURAZYME infusions in Study 2 were anaphylactic and hypersensitivity reactions (see Precautions). The most common adverse reactions requiring intervention were infusion reactions reported in 49% (22 of 45) of patients treated with ALDURAZYME. The most commonly reported infusion reactions included rash (13%), flushing (11%), pyrexia (11%), headache (9%), abdominal pain or discomfort (9%), and injection site reaction (9%). Less commonly reported infusion reactions included nausea (7%), diarrhea (7%), feeling hot or cold (7%), vomiting (4%), pruritus (4%), arthralgia (4%), and urticaria (4%). Additional common adverse reactions included back pain and musculoskeletal pain.
Clinical Trials in Patients 6 Years and Younger: Study 3 was a 52-week, open-label, uncontrolled study of 20 MPS I patients, ages 6 months to 5 years old (at enrollment). Sixteen patients were clinically assessed as having the Hurler form, and 4 had the Hurler-Scheie form. All 20 patients received ALDURAZYME at 0.58 mg/kg of body weight once weekly for 26 weeks and up to 52 weeks. All patients were treated with antipyretics and antihistamines prior to the infusions.
The most commonly reported serious adverse events (regardless of relationship) reported with ALDURAZYME infusions in Study 3 were otitis media (20%), and central venous catheterization required for ALDURAZYME infusion (15%).
The nature and severity of infusion reactions were similar between the older and less severely affected patients in Studies 1 and 2, and the younger, more severely affected patients in Study 3. The most commonly reported adverse reactions in Study 3 were infusion reactions reported in 35% (7 of 20) of patients and included pyrexia (30%), chills (20%), blood pressure increased (10%), tachycardia (10%), and oxygen saturation decreased (10%). Other commonly reported infusion reactions occurring in ≥ 5% of patients were pallor, tremor, respiratory distress, wheezing, crepitations (pulmonary), pruritus, and rash.
Immunogenicity: As with all the therapeutic proteins, there is potential for immunogenicity. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described as follows with the incidence of antibodies in other studies or to other to laronidase products may be misleading.
In clinical trials, 99 of 102 patients (97%) treated with ALDURAZYME were positive for IgG antibodies to ALDURAZYME. No correlation was demonstrated between the presence of IgG anti-ALDURAZYME antibodies and therapeutic response (6 MWT and FVC) or the occurrence of hypersensitivity reactions. Potential for antibody neutralization of cellular uptake has not been assessed. No consistent association was demonstrated between the presence of antibodies that neutralize enzymatic activity and therapeutic response.
The data reflect the percentage of patients whose test results were considered positive for antibodies to ALDURAZYME using a specific enzyme-linked immunosorbent assay (ELISA) and confirmed by radio-immunoprecipitation (RIP). ALDURAZYME IgG antibodies were reported as titers. Drug specific antibody was detected in 42 of the 45 patients (93.3%) treated in Study 1 and Study 2. The mean time to seroconversion was 51 days in patients 6 years and older. In Study 3, all patients (100%) 5 years old or younger developed IgG antibodies against ALDURAZYME with a mean time to seroconversion of 26 days [see Pharmacology: Pharmacodynamics: Clinical Studies under Actions].
Nine patients in Study 1 and Study 2, collectively, who experienced severe infusion reactions were tested for ALDURAZYME-specific IgE antibodies and complement activation. IgE testing was performed by ELISA, and complement activation was measured by the Quidel Enzyme Immunoassay. One of the nine patients had an anaphylactic reaction consisting of urticaria and airway obstruction and tested positive for both ALDURAZYME-specific IgE binding antibodies and complement activation. None of the patients in the open-label clinical study of patients 5 years old or younger (Study 3) tested positive for IgE.;
Other hypersensitivity reactions were also seen in patients receiving ALDURAZYME (see Adverse Reactions).
In the post-marketing setting, approximately 1% of patients experienced severe or serious infusion hypersensitivity reactions and tested positive for IgE. Of these IgE-positive patients, some have discontinued treatment, but some have been successfully re-challenged. The clinical significance of IgE antibodies has not been established.
Post-marketing Experience: The following adverse reactions have been identified during post approval use of ALDURAZYME. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In post-marketing experience with ALDURAZYME, severe and serious infusion reactions have been reported, some of which were life-threatening, including anaphylactic shock (see Warnings and Precautions) and laryngeal oedema.
Adverse reactions resulting in death reported in the post-marketing setting with ALDURAZYME treatment included cardiorespiratory arrest, respiratory failure, cardiac failure, and pneumonia. These events have been reported in MPS I patients with significant underlying disease.
Additional adverse reactions included fatigue, oedema peripheral, erythema and cyanosis.
There have been a small number of reports of extravasation in patients treated with ALDURAZYME. There have been no reports of tissue necrosis associated with extravasation.
To report SUSPECTED ADVERSE REACTIONS, please email to Med.SAMS@sanofi.com


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
