Adverse reactions for CML: Infections and infestations: Herpes zoster, herpes simplex, nasopharyngitis, pneumonia
1, sinusitis, cellulitis, upper respiratory tract infection, influenza, urinary tract infection, gastroenteritis, sepsis and fungal infection.
Blood and lymphatic system disorders: Neutropenia, thrombocytopenia, anaemia, pancytopenia, febrile neutropenia, thrombocytopenia, lymphopenia, bone marrow depression, eosinophilia, lymphadenopathy, haemolytic anaemia.
Metabolism and nutrition disorder: Anorexia, hypokalemia, increased appetite, hypophosphataemia, decreased appetite, dehydration, gout, hyperuricemia, hypercalcaemia, hyperglycaemia, hypernatremia, hyperkalaemia, hypomagnesaemia.
Psychiatric disorders: Insomnia, depression, libido decreased, anxiety and confusional state.
Nervous system disorders: Dizziness, paraesthesia, taste disturbance, hypoaesthesia, migraine, somnolence, syncope, peripheral neuropathy, memory impairment, sciatica, restless leg syndrome, tremor, cerebral haemorrhage, increased intracranial pressure, convulsions, optic neuritis.
Eye disorders: Eyelid oedema, lacrimation increased, conjunctival haemorrhage, conjunctivitis, dry eye, blurred vision, eye irritation, eye pain, orbital oedema, scleral haemorrhage, retinal haemorrhage, blepharitis, macular oedema, cataract, glaucoma, papilloedema.
Ear and labyrinth disorders: Vertigo, tinnitus, hearing loss.
Cardiac disorders: Palpitations, tachycardia, cardiac failure congestive
2, pulmonary oedema, arrhythmia, atrial fibrillation, cardiac arrest, myocardial infarction, angina pectoris, pericardial effusion.
Vascular disorders3: Flushing, haemorrhage, hypertension, haematoma, subdural hematoma, peripheral coldness, hypotension, Raynaud's phenomenon.
Respiratory, thoracic and mediastinal disorders: Dyspnoea, epistaxis, cough, pleural effusion
4, pharyngolaryngeal pain, pharyngitis, pleuritic pain, pulmonary fibrosis, pulmonary hypertension, pulmonary haemorrhage.
Gastrointestinal disorders: Nausea, diarrhoea, vomiting, dyspepsia, flatulence, abdominal distension, gastro-oesophageal reflux, constipation, dry mouth, gastritis, stomatitis, mouth ulceration, eructation, melaena, oesophagitis, ascites, gastric ulcer, haematemesis, cheilitis, dysphagia, pancreatitis, colitis, inflammatory bowel disease, ileus.
Hepatobiliary disorders: Increased hepatic enzymes, hyperbilirubinemia, hepatitis, jaundice, hepatic failure
6, hepatic necrosis
6.
Skin and subcutaneous tissue disorders: Periorbital oedema, dermatitis/eczema/rash, pruritus, face oedema, dry skin, erythema, alopecia, night sweats, photosensitivity reaction, rash pustular, contusion, sweating increased, urticaria, ecchymosis, increased tendency to bruise, hypotrichosis, skin hypopigmentation, dermatitis exfoliative, onychoclasis, folliculitis, petechiae, psoriasis, purpura, skin hyperpigmentation, bullous eruption, acute febrile neutrophilic dermatosis (Sweet's syndrome), nail discolouration, angioneurotic oedema, rash vesicular, erythema multiforme, leucocytoclastic vasculitis, Stevens-johnson syndrome, acute generalised exanthematous pustulosis (AGEP).
Musculoskeletal and connective tissue disorders: Muscle spasm and cramps, musculoskeletal pain including myalgia, arthralgia, bone pain
5, joint swelling, joint and muscle stiffness, muscular weakness, arthritis.
Renal and urinary disorders: Renal pain, haematuria, renal failure acute, urinary frequency increased.
Reproductive system and breast disorders: Gynaecomastia, erectile dysfunction, menorrhagia, menstruation irregular, sexual dysfunction, nipple pain, breast enlargement, scrotal oedema.
General disorders and administration site conditions: Fluid retention and oedema, fatigue, weakness, pyrexia, anasarca, chills, rigors, chest pain, malaise.
Investigations: Weight increased, weight decreased, blood creatinine increased, blood creatine phosphokinase increased, blood lactate dehydrogenase increased, blood alkaline phosphatase increased, blood amylase increased.
Note: 1 Pneumonia was reported in patients with transformed CML.
2 Cardiac events including congestive heart failure were observed in patients with transformed CML than in patients with chronic CML.
3 Bleeding (haematoma, haemorrhage) was reported in patients with transformed CML (CML-AP and CML-BC).
4 Pleural effusion can be occur in patients with transformed CML (CML-AP and CML-BC) than in patients with chronic CML.
5 Musculoskeletal pain and related events can be occur in patients with CML.
6 Some fatal cases of hepatic failure and hepatic necrosis can be occur.
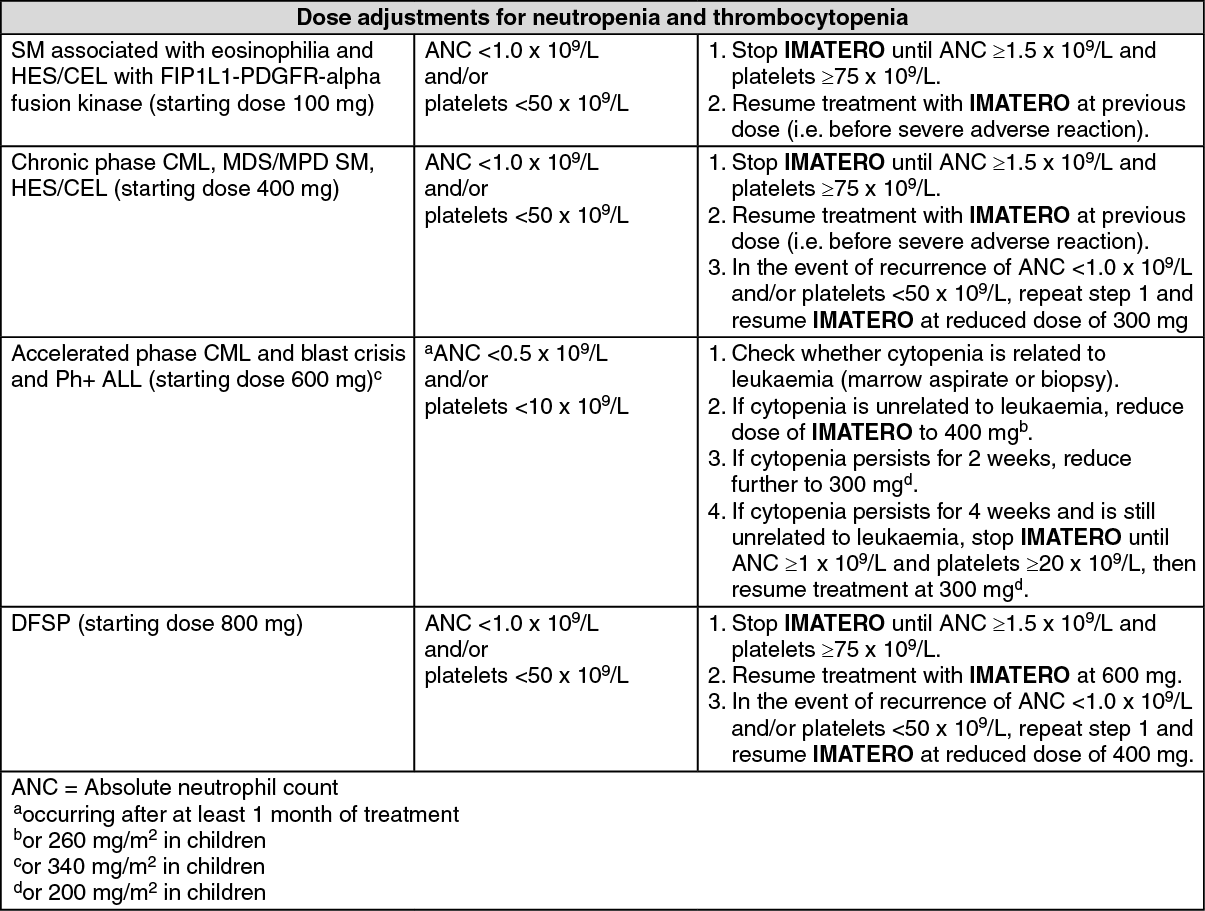
Description of selected adverse drug reactions: Myelosuppression: Myelosuppression is reported in cancer patients treated with Imatinib. Myelosuppression, thrombocytopenia, neutropenia and anemia were reported grade 3 and 4 laboratory abnormalities. Overall, the myelosuppression experienced with Imatinib in CML patient was generally reversible and in most patients did not result in dose interruption or doe reduction. Few patients required drug discontinuation. Other events of pancytopenia, lymphopenia and bone marrow depression have also been reported.
Hematologic depression appeared greatest at the highest doses and also appeared to the dependent on stage of CML disease, with grade 3 or 4 neutropenia and thrombocytopenia between 4 and 6 times higher in blast and accelerated phase as compared to newly diagnosed patients in CP CML.
These events can usually be managed with either a reduction of the dose or an interruption of treatment with Imatinib, but require discontinuation. The incidence of hematologic toxicities is less in patients with solid tumors than in patients with Ph+ leukemias, with Grade
¾ neutropenia and thrombocytopenia occurring approximately 10% and 1% respectively.
Hemorrhage: CNS and GI hemorrhages are not uncommon in CML patients with compromised marrow function at baseline. Hemorrhages are well-recognized part of the disease complications in an acutely ill population of leukemia patients and may result from thrombocytopenia, or less commonly, platelet dysfunction. However, not all patients experiencing CNS and GI hemorrhages during therapy with Imatinib are thrombocytopenic.
The most common manifestation of clinically significant bleeding was GI haemorrhage, which occurred most commonly in advanced CML patients where bleeding might occur as part of the underlying disease due to tumor bleeding from tumor haemorrhage/tumor necrosis. In first line CML, the observed frequencies of GI haemorrhage were generally the lowest. Gastric antral vascular ectasia (GAVE) is also rarely occur with Imatinib use.
Edema and fluid retention: Edema is a common toxicity of Imatinib appearing in greater than 50% of all patients across all indications. Edema is dose-related and there appears to be a correlation with its occurrence and plasma levels. The most common manifestations is periorbital edema and lower extremity edema. Specific therapy is not usually required. Other fluid retention events occur much less, but due to the location of the anatomic site may be potentially serious. The most frequent fluid retention event was pleural effusion, most commonly observed in advanced CML. The frequency of cardiac failure was generally low in patients with edema and fluid retention. It was higher in advanced CML than in other groups. This could be explained by the worse medical condition of advanced patients. The same trend was observed for renal failure in patients with edema and fluid retention. Most patients with edema and fluid retention were elderly (> 65 years old).
The frequency of events suggesting congestive heart failure was appreciably higher in patients with transformed CML (accelerated phase or blast crisis), higher age or with baseline haemoglobin of less than 8g/dL. Congestive heart failure (CHF) and left ventricular dysfunction have since been continuously monitored in the PSUR.
Skin rashes and severe cutaneous adverse reactions: A generalized erythematous, maculopapular, pruritic skin rash that can fade despite continued therapy, has been reported. Some patients may have pruritus without accompanying rash, and sometimes there is an exfoliative component. Re-exposure in some patients has resulted in reappearance of rash, but not in all patients. These eruptions generally respond to antihistamines and topical steroids. Occasionally, systemic steroids are required.
Skin rashes have been observed in patients treated with Imatinib across all indications. These are frequently pruritic and most commonly appear as erythematous, maculopapular lesions on the forearm, the trunk or the face. Skin biopsies have revealed a toxic drug reaction with a mixed cellular infiltrate. Although most rashes are mild and self limiting, more severe cases may require interruption or discontinuation of treatment.
Hepatotoxicity: Hepatotoxicity, occasionally severe, may occur, and has been observed. LFT abnormalities usually consisted of mild elevations in transaminases, although a minority of patients had elevated levels of bilirubin. Onset is generally within the first two months of therapy, but has occurred as late as 6 to 12 months after commencing therapy. The levels generally normalize after withholding therapy for 1 to 4 weeks.
Hypophosphatemia: Low serum phosphate and hypophosphatemia (up to Grade
¾) can be occur relatively commonly across all indications, however the origin and the significance of this finding have not been established. Imatinib has been shown to inhibit the differentiation of human monocytes into osteoclasts. The decrease was accompanied by a decrease in the resorptive capacity of these cells. A dose-dependent decrease of RANK-L was observed in osteoclasts in the presence of Imatinib. Sustained inhibition of osteoclastic activity may lead to counter regulatory response resulting in increased levels of PTH. The clinical relevance of the preclinical findings is yet unclear and an association with skeletal AEs such as bone fractures has not been demonstrated.
Gastrointestinal obstruction, perforation or ulceration: GI ulceration, which can be occur local irritation by Imatinib, has been occur in a small proportion of patient. Tumor haemorrhage/tumor necrosis, obstruction and GI perforation seem to be disease-related.
Tumor lysis syndrome: A causal relationship between tumor lysis syndrome and Imatinib treatment is deemed possible, although some cases were confounded by concomitant medications and other independent risks (see PRECAUTIONS).
Growth retardation in children: Imatinib appears to affect the stature of children, especially children who are pre-pubertal. A causal relationship between growth retardation in children and Imatinib treatment could not be ruled out although for some cases of growth retardation there was limited information.
Severe respiratory adverse drug reaction: Severe respiratory events, sometimes fatal, have been observed with Imatinib treatment, including acute respiratory failure, pulmonary hypertension, interstitial lung disease and pulmonary fibrosis. Pre-existing cardiac or pulmonary conditions that may be associated with severe respiratory events can be occur.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
