Pharmacotherapeutic group: Medicinal products for treatment of bone diseases, medicinal products affecting bone structure and mineralization.
ATC code: M05BX06.
Pharmacology: Pharmacodynamics: Mechanism of action: Romosozumab is a humanized monoclonal antibody (IgG2) that binds and inhibits sclerostin, thereby increasing bone formation due to the activation of bone lining cells, increasing bone matrix production by osteoblasts, and recruitment of osteoprogenitor cells. Additionally, romosozumab results in changes to expression of osteoclast mediators, thereby decreasing bone resorption. Together, this dual effect of increasing bone formation and decreasing bone resorption results in rapid increases in trabecular and cortical bone mass, improvements in bone structure, and strength.
Pharmacodynamic effects: In postmenopausal women with osteoporosis, romosozumab increased the bone formation marker procollagen Type 1 N terminal propeptide (P1NP) early in treatment, with a peak increase of approximately 145% relative to placebo 2 weeks after initiating treatment, followed by a return to placebo levels at month 9 and a decline to approximately 15% below placebo at month 12. Romosozumab decreased the bone resorption marker type-1 collagen C-telopeptide (CTX) with a maximal reduction of approximately 55% relative to placebo 2 weeks after initiating treatment. CTX levels remained below placebo and were approximately 25% below placebo at month 12.
After discontinuation of romosozumab therapy in postmenopausal women with osteoporosis, P1NP levels returned to baseline within 12 months; CTX increased above baseline levels within 3 months and returned toward baseline levels by month 12, reflecting reversibility of effect. Upon retreatment with romosozumab (in a limited number of patients) after 12 months placebo treatment, the levels of increase in P1NP and decrease in CTX by romosozumab were similar to that observed during the initial treatment.
Clinical trial efficacy: Treatment of osteoporosis in postmenopausal women: Efficacy and safety of romosozumab was assessed in two pivotal studies, an alendronate-controlled (ARCH) and a placebo-controlled study (FRAME).
Study 20110142 (ARCH): The efficacy and safety of romosozumab in the treatment of osteoporosis in postmenopausal women was evaluated in a multicenter, multinational, randomized, double-blind, alendronate-controlled, superiority study of 4,093 postmenopausal women aged 55 to 90 years (mean age of 74.3 years) with previous fragility fractures.
Enrolled women had either a BMD (Bone Mineral Density) T-score at the total hip or femoral neck of ≤ -2.50, and either at least 1 moderate or severe vertebral fracture; or at least 2 mild vertebral fractures; or a BMD T-score at the total hip or femoral neck of ≤ -2.00, and either at least 2 moderate or severe vertebral fractures; or a fracture of the proximal femur that occurred within 3 to 24 months prior to randomization.
The mean baseline lumbar spine, total hip, and femoral neck BMD T-scores were -2.96, -2.80, and -2.90, respectively, 96.1% of women had a vertebral fracture at baseline, and 99.0% of women had a previous osteoporotic fracture. Women were randomized (1:1) to receive either monthly subcutaneous injections of romosozumab or oral weekly alendronate in a blinded fashion for 12 months. After the 12-month double blind study period, women in both arms transitioned to alendronate while remaining blinded to their initial treatment. The primary analysis was performed when all women had completed the month 24 study visit and clinical fracture events were confirmed for at least 330 women and occurred after a median follow-up time of approximately 33 months on study. Women received calcium and vitamin D supplementation daily.
The primary efficacy endpoints were the incidence of new vertebral fracture through month 24 and the incidence of clinical fracture (nonvertebral fracture and clinical vertebral fracture) at primary analysis.
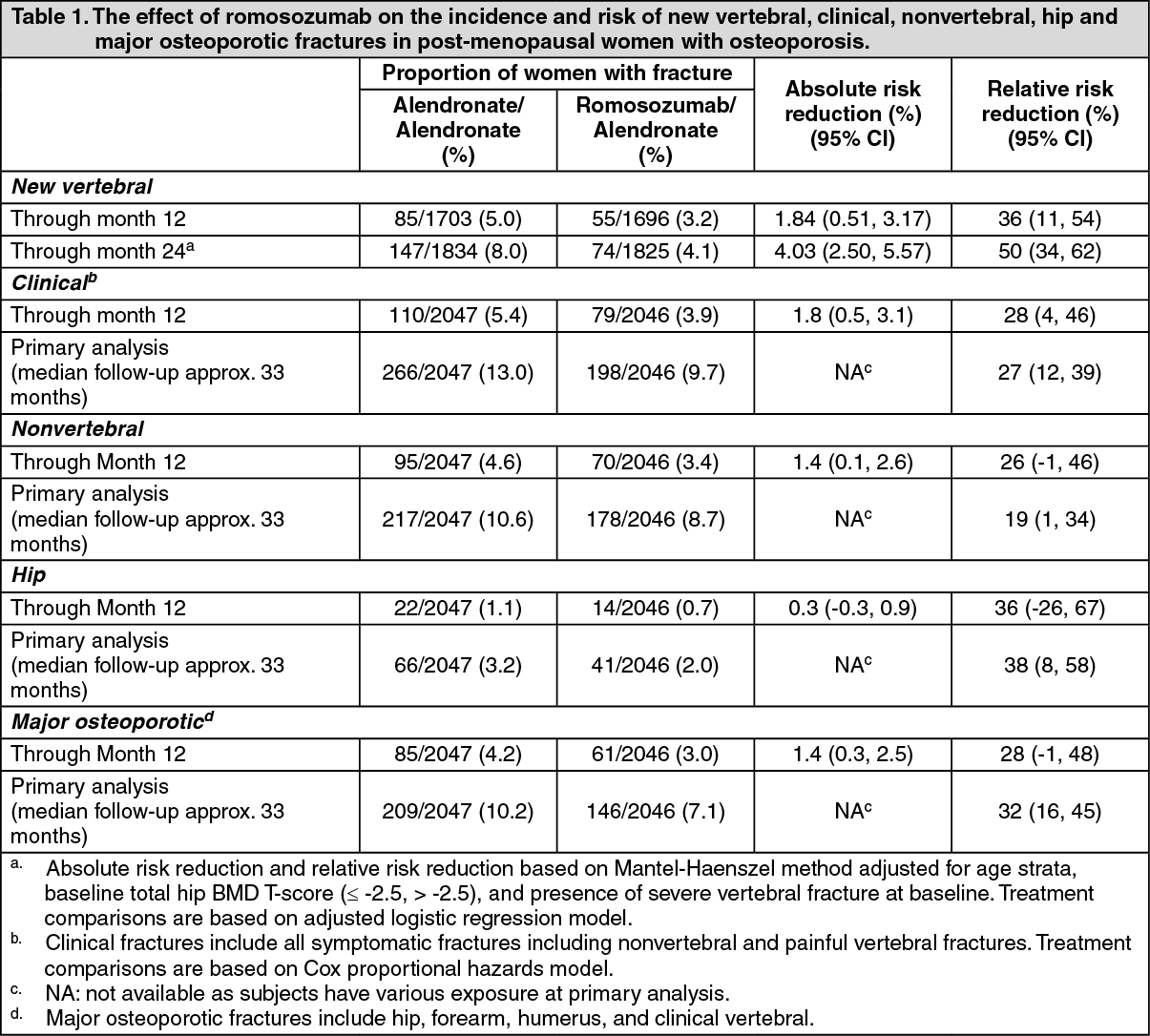
Effect on new vertebral, clinical, nonvertebral, hip and major osteoporotic fractures: As shown in Table 1, romosozumab reduced the incidence of new vertebral fracture through month 24 (adjusted p-value < 0.001) and the incidence of clinical fracture at primary analysis (adjusted p-value < 0.001) as well as the incidence of non vertebral fractures at primary analysis (adjusted p-value = 0.040) versus treatment with alendronate alone. Table 1 also shows nonvertebral, hip and major osteoporotic fracture risk reduction through primary analysis, month 12 and month 24. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Effect on Bone Mineral Density (BMD): In postmenopausal women with osteoporosis, romosozumab for 12 months followed by alendronate for 12 months increased BMD compared with alendronate alone at month 12 and 24 (p-value < 0.001) (see Table 2).
Following 12 months of treatment, romosozumab increased BMD at the lumbar spine from baseline in 98% of postmenopausal women. (See Table 2.)
Click on icon to see table/diagram/image
The significant difference in BMD achieved in the first 12 months was maintained through month 36 upon transition/continuation to alendronate. Treatment differences were observed at 6 months at lumbar spine, total hip and femoral neck.
Study 20070337 (FRAME): The efficacy and safety of romosozumab in the treatment of postmenopausal osteoporosis was evaluated in a multicenter, multinational, randomized, double-blind, placebo-controlled, parallel-group study of 7,180 postmenopausal women aged 55 to 90 years (mean age of 70.9 years). 40.8% of enrolled women had severe osteoporosis with a prior fracture at baseline.
The co-primary efficacy endpoints were the incidence of new vertebral fractures through month 12 and through month 24.
Romosozumab reduced the incidence of new vertebral fractures through month 12 (absolute risk reduction: 1.3% [95% CI: 0.79; 1.80], relative risk reduction: 73% [95% CI: 53; 84], adjusted p-value < 0.001) and after transition to denosumab through month 24 (absolute risk reduction: 1.89 % [95% CI: 1.30; 2.49], relative risk reduction: 75% [95% CI: 60; 84], adjusted p-value < 0.001).
Women transitioning from bisphosphonate therapy: Study 20080289 (STRUCTURE): The safety and efficacy of romosozumab in postmenopausal women with severe osteoporosis transitioning from bisphosphonate therapy (92.7% in teriparatide group and 88.1% in romosozumab group had prior alendronate use during the last 3 years) were evaluated in a multicenter, randomized, open-label study of 436 postmenopausal women aged 56 to 90 years (mean age of 71.5 years) versus teriparatide.
The primary efficacy variable was percent change in total hip BMD from baseline at month 12. Romosozumab significantly increased BMD at the total hip relative to teriparatide at month 12 (mean treatment difference from Teriparatide: 3.4% [95% CI: 2.8; 4.0], p-value < 0.0001). The trial was not intended to estimate the effect on fractures but there were seven fractures in the romosozumab arm and nine fractures in the teriparatide arm of the study.
Bone Histology and Histomorphometry: In a bone histology sub-study, a total of 154 transiliac crest bone biopsy specimens were obtained from 139 postmenopausal women with osteoporosis at months 2 and 12 (in FRAME study).
Qualitative histology assessments showed normal bone architecture and quality at all time points, normal lamellar bone with no evidence of mineralization defects, woven bone, marrow fibrosis, or clinically significant marrow abnormality in patients treated with romosozumab.
Histomorphometry assessments on biopsies at months 2 and 12 in women showed an increase of bone formation parameters and a decrease in bone resorption parameters while bone volume and trabecular thickness were increased in romosozumab group compared to placebo group.
Paediatric population: See Dosage & Administration for information on paediatric use.
Pharmacokinetics: Absorption: The median time to maximum romosozumab concentration (t
max) was 5 days (range: 2 to 7 days). Following a 210 mg subcutaneous dose, bioavailability was 81%.
Biotransformation: Romosozumab is a humanized monoclonal antibody (IgG2) with high affinity and specificity for sclerostin, and therefore is cleared via a rapid saturable elimination pathway (i.e. target mediated nonlinear clearance, mediated by degradation of the romosozumab-sclerostin complex) and via a slow nonspecific elimination pathway mediated by the reticuloendothelial system.
Elimination: After C
max, serum levels declined with a mean effective half-life of 12.8 days. Steady-state was generally reached by month 3 with less than 2-fold accumulation following monthly dosing.
Linearity/non-linearity: Following subcutaneous administration, romosozumab exhibits non-linear pharmacokinetics as a result of binding to sclerostin. Multiple doses administered ranged from 70 to 210 mg.
Renal impairment: Following a 210 mg dose of romosozumab in a clinical trial of 16 patients with severe renal impairment (creatinine clearance < 30 ml/min) or end-stage renal disease (ESRD) receiving haemodialysis, mean C
max and AUC were 29% and 44% higher in patients with severe renal impairment as compared to healthy subjects. Mean romosozumab exposure was similar in patients with ESRD receiving haemodialysis as compared to healthy subjects.
Population pharmacokinetic analysis indicated an increase in romosozumab exposure with increasing severity of renal impairment. However, based on an exposure-response model of BMD changes and comparison to exposures obtained at tolerated clinical doses, no dose adjustment is recommended in these patients. Monitoring of hypocalcemia in patients with severe renal impairment or receiving dialysis is recommended (see Precautions).
Hepatic impairment: No clinical trials have been conducted to evaluate the effect of hepatic impairment. Hepatic impairment is not expected to impact on the pharmacokinetics of romosozumab since the liver is not a major organ for romosozumab metabolism or excretion.
Elderly: The pharmacokinetics of romosozumab were not affected by age from 20 years to 89 years.
Bodyweight: Romosozumab exposure decreased with increasing body weight however this decrease had a minimal impact on lumbar spine BMD gain based on exposure-response analysis and is not clinically meaningful. Based on population PK analyses, the expected median steady state AUC for a 61 kg and 114 kg patient is 558 μg·day/ml and 276 μg·day/ml respectively following a monthly subcutaneous dose of 210 mg romosozumab.
Ethnicity and gender: No dose adjustment is necessary for any patient characteristics. Based on a population pharmacokinetic analysis, gender and race (Japanese versus non-Japanese) had no clinically meaningful impact on the pharmacokinetics of romosozumab (< 20% change in exposure at steady state).
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, carcinogenic potential or in bone safety studies.
In a carcinogenicity study, doses up to 50 mg/kg/week were administered by subcutaneous injection to Sprague-Dawley male and female rats from 8 weeks of age for up to 98 weeks. These doses resulted in systemic exposures that were up to 19 times higher than the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg romosozumab (based on AUC comparison).
Romosozumab caused a dose-dependent increase in bone mass with macroscopic bone thickening at all doses. There were no effects of romosozumab on mortality or tumor incidence in male or female rats.
Studies in female and male rats did not show any romosozumab-related effects on mating, fertility, or male reproductive assessments (sperm parameters or organ weights), and there were no effects on estrous cycling or any ovarian or uterine parameters at exposures around 54 times the clinical exposure.
Skeletal malformations, including syndactyly and polydactyly, were observed at a low incidence in 1 out of 75 litters at exposures around 30 times the clinical exposure following administration of romosozumab to rats during the period of organogenesis. There were no adverse effects on postnatal growth and development.
Sclerostin has been suggested to have a role in digit formation, however, as digit formation in the human occurs in the first trimester when placental transfer of immunoglobulins is limited, the risk of a similar finding in humans is low (see Use in Pregnancy & Lactation).



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image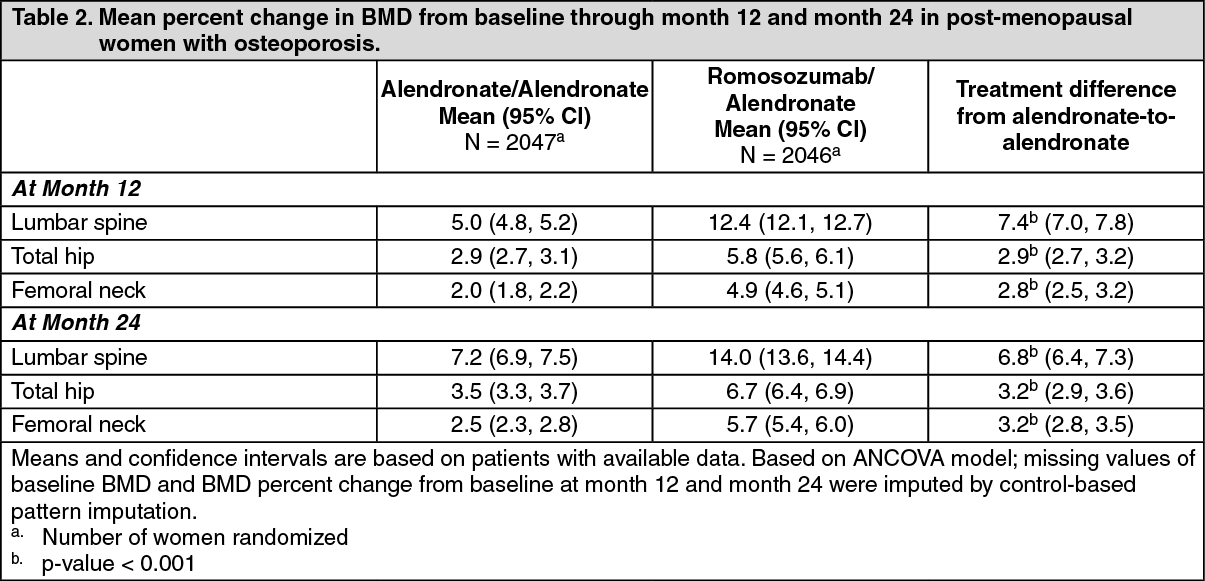 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
