Nhóm dược lý: Emicizumab là kháng thể đơn dòng, nguồn gốc từ người, bản chất là immunoglobulin G4 (IgG4) biến đổi, có cấu trúc đặc biệt gắn với yếu tố IXa và yếu tố X, được tạo thành bằng kỹ thuật ADN tái tổ hợp trong tế bào buồng trứng chuột lang Trung quốc.
Mã ATC: B02BX06
ĐẶC TÍNH DƯỢC LỰC HỌC
Cơ chế tác dụng
Emicizumab tạo cầu nối cho yếu tố IX hoạt hóa và yếu tố X để khôi phục lại chức năng thiếu hụt yếu tố VIII hoạt hóa cần thiết cho quá trình đông máu.
Emicizumab không có mối liên quan về mặt cấu trúc hoặc tương đồng về trình tự với yếu tố VIII, do đó không gây ra hoặc tăng tạo các chất ức chế trực tiếp yếu tố VIII.
Dược lực học
Hemophilia A là một bệnh rối loạn đông máu di truyền liên quan đến nhiễm sắc thể X do thiếu hụt chức năng yếu tố VIII dẫn đến chảy máu trong khớp, cơ hoặc nội tạng, tự phát hoặc do tai nạn, chấn thương hay phẫu thuật. Điều trị dự phòng bằng HEMLIBRA đã được ghi nhận giúp rút ngắn thời gian aPTT và làm tăng hoạt tính của yếu tố VIII (sử dụng xét nghiệm chromogen với các yếu tố đông máu ở người). Hai chất chỉ điểm dược lực học này không phản ánh tác dụng chống đông máu
in vivo thực tế của emicizumab (aPTT bị rút ngắn quá mức và hoạt tính của yếu tố VIII được ghi nhận quá mức) nhưng phản ánh tương đối về tác dụng tiền đông máu của emicizumab.
Nghiên cứu lâm sàng/hiệu quả
Hiệu quả của HEMLIBRA trong điều trị dự phòng thường quy ở bệnh nhân mắc hemophilia A có hoặc không có chất ức chế được đánh giá trong 4 nghiên cứu lâm sàng (3 nghiên cứu trên người lớn và thiếu niên [HAVEN 3, HAVEN 1, và HAVEN 4] và một nghiên cứu trên trẻ em [HAVEN 2]).
Các nghiên cứu lâm sàng trên bệnh nhân người lớn và thiếu niên
• HAVEN 3
Nghiên cứu HAVEN 3 là nghiên cứu lâm sàng pha III ngẫu nhiên, đa trung tâm, nhãn mở trên 152 nam giới người lớn và thiếu niên (từ 12 tuổi trở lên và nặng > 40 kg) mắc hemophilia A không có chất ức chế yếu tố VIII trước đó đã điều trị từng đợt (“theo yêu cầu”) hoặc điều trị dự phòng với yếu tố VIII. Bệnh nhân được dùng HEMLIBRA tiêm dưới da, 3 mg/kg 1 lần/tuần trong 4 tuần đầu sau đó là 1,5 mg/kg 1 lần/tuần (nhóm A và D) hoặc 3 mg/kg mỗi 2 tuần (nhóm B), hoặc không dự phòng (nhóm C). Bệnh nhân ở nhóm C có thể chuyển sang dùng HEMLIBRA (3 mg/kg mỗi 2 tuần) sau khi hoàn thành ít nhất 24 tuần không điều trị dự phòng. Đối với nhóm A và B liều được cho phép điều chỉnh lên tới 3 mg/kg mỗi tuần sau 24 tuần ở những bệnh nhân trải qua chảy máu đủ điều kiện (ví dụ như, chảy máu tự phát và chảy máu lâm sàng đáng kể xảy ra ở tình trạng ổn định) từ 2 lần trở lên. Bệnh nhân ở nhóm D có thể điều chỉnh tăng liều sau lần thứ 2 chảy máu đủ điều kiện. Tại thời điểm phân tích tạm thời, năm bệnh nhân được điều chỉnh tăng liều ở liều điều trị duy trì của họ.
89 bệnh nhân trước đó đã điều trị yếu tố VIII từng đợt (“theo yêu cầu”) được phân ngẫu nhiên theo tỷ lệ 2:2:1 để dùng HEMLIBRA 1 lần mỗi tuần (nhóm A; N=36), mỗi 2 tuần (nhóm B; N=35) hoặc không dự phòng (nhóm C; N=18), với sự phân tầng theo tỷ lệ chảy máu trước 24 tuần (< 9 hoặc ≥ 9). 63 bệnh nhân được điều trị yếu tố VIII dự phòng trước đó được đưa vào nhóm D để nhận HEMLIBRA (1,5 mg/kg 1 lần/tuần).
Mục tiêu chính của nghiên cứu là đánh giá hiệu quả của điều trị duy trì HEMLIBRA hàng tuần (nhóm A) và mỗi 2 tuần (nhóm B) so với không duy trì (nhóm C) ở những bệnh nhân trước đó được điều trị từng đợt với yếu tố VIII dựa trên số lần chảy máu cần điều trị với yếu tố đông máu (xem bảng 1). Các mục tiêu khác của nghiên cứu bao gồm đánh giá so sánh ngẫu nhiên của nhóm A hoặc B với nhóm C về hiệu quả của dự phòng với HEMLIBRA trong giảm tất cả số lần chảy máu, chảy máu tự phát, chảy máu khớp và target chảy máu khớp đích (xem bảng 2), cũng như đánh giá chất lượng cuộc sống liên quan đến sức khỏe của bệnh nhân (xem bảng 9). Lựa chọn điều trị bệnh nhân cũng được sử dụng đánh giá bằng cách sử dụng khảo sát ưu tiên.
• HAVEN 1
Nghiên cứu HAVEN 1 là 1 nghiên cứu lâm sàng ngẫu nhiên, đa trung tâm, nhãn mở trên 109 bệnh nhân nam người lớn và thiếu niên (từ 12 tuổi trở lên và nặng trên 40 kg) bị hemophilia A có chất ức chế yếu tố VIII, trước đây đã từng được điều trị theo đợt (“theo yêu cầu”) hoặc điều trị dự phòng bằng thuốc khác. Trong nghiên cứu, bệnh nhân được điều trị dự phòng hàng tuần bằng HEMLIBRA (Nhóm A, C và D) - liều 3 mg/kg, 1 lần/tuần trong 4 tuần, sau đó 1,5 mg/kg 1 lần/tuần - hoặc không điều trị dự phòng (Nhóm B). Bệnh nhân được phân ngẫu nhiên vào nhóm B có thể chuyển sang điều trị dự phòng bằng HEMLIBRA sau ít nhất 24 tuần không điều trị dự phòng. Được phép tăng liều đến 3 mg/kg mỗi tuần sau 24 tuần điều trị dự phòng bằng HEMLIBRA cho những bệnh nhân đã trải qua từ 2 đợt chảy máu đạt yêu cầu trở lên (như có ≥ 2 lần chảy máu tự phát xảy ra ở trạng thái ổn định, có ý nghĩa lâm sàng). Trong nghiên cứu, 2 bệnh nhân được điều chỉnh liều duy trì đến 3 mg/kg, 1 lần/tuần.
53 bệnh nhân trước đây đã điều trị bằng thuốc khác theo đợt (“theo yêu cầu”) được phân nhóm ngẫu nhiên với tỷ lệ 2:1 được điều trị dự phòng bằng HEMLIBRA (nhóm A) hoặc không điều trị dự phòng (nhóm B) với sự phân tầng theo tỷ lệ bị chảy máu trong 24 tuần (< 9 hoặc ≥ 9).
49 bệnh nhân trước đây được điều trị dự phòng bằng các thuốc dự phòng khác tham gia vào nhóm C điều trị dự phòng bằng HEMLIBRA. 7 bệnh nhân trước đây được điều trị bằng thuốc khác từng đợt (“theo yêu cầu”), đã tham gia vào nghiên cứu không can thiệp trước đó, nhưng không được tham gia vào HAVEN 1 trước giai đoạn tuyển chọn nhóm A và B kết thúc, có thể tham gia vào nhóm D dự phòng bằng HEMLIBRA.
Mục tiêu chính của nghiên cứu là đánh giá hiệu quả điều trị dự phòng bằng HEMLIBRA hàng tuần trên các bệnh nhân trước đây đã được điều trị dự phòng bằng các thuốc khác (“theo yêu cầu”) so với không điều trị dự phòng (Nhóm A so với Nhóm B) dựa trên số lần chảy máu cần phải điều trị bằng yếu tô đông máu theo thời gian (ít nhất 24 tuần hoặc ngày ngừng điều trị) (xem bảng 1). Các mục tiêu phụ khác của việc so sánh ngẫu nhiên nhóm A và B là hiệu quả điều trị dự phòng hàng tuần của HEMLIBRA trong việc làm giảm số lần chảy máu, chảy máu tự phát, chảy máu ở khớp và khớp đích (xem bảng 4), cũng như đánh giá chất lượng cuộc sống và sức khỏe của bệnh nhân (xem bảng 9 và 10). Hiệu quả điều trị dự phòng hàng tuần của HEMLIBRA so sánh với điều trị với các thuốc dự phòng khác trước đây cũng được đánh giá trên bệnh nhân đã tham gia nghiên cứu không can thiệp trước đó (các nhóm) (xem bảng 5). Chỉ có các bệnh nhân từ nghiên cứu không can thiệp được đưa vào so sánh này do dữ liệu về chảy máu và điều trị được thu thập chi tiết cùng mức độ trong nghiên cứu như đã được sử dụng trong HAVEN 1.
• HAVEN 4
HEMLIBRA đã được nghiên cứu trong một nghiên cứu lâm sàng pha III đơn nhóm, đa trung tâm trên 41 nam giới trưởng thành và thiếu niên (từ 12 tuổi trở lên và nặng trên 40 kg) mắc hemophilia A có hoặc không có chất ức chế yếu tố 8 mà trước đó đã được điều trị từng đợt (“theo yêu cầu”) hoặc điều trị dự phòng với các thuốc dự phòng khác hoặc với yếu tố VIII. Bệnh nhân sử dụng HEMLIBRA dự phòng với liều 3 mg/kg 1 lần/tuần trong 4 tuần sau đó là 6 mg/kg mỗi 4 tuần.
Mục tiêu chính của nghiên cứu là đánh giá hiệu quả của dự phòng bằng HEMLIBRA trong việc duy trì kiểm soát chảy máu đầy đủ, cứ 4 tuần một lần dựa trên chảy máu được điều trị. Các mục tiêu khác của nghiên cứu là đánh giá hiệu quả lâm sàng của dự phòng bằng HEMLIBRA trên tất cả các chảy máu, chảy máu tự phát có điều trị, chảy máu khớp và khớp đích có điều trị (xem bảng 7). Lựa chọn điều trị bệnh nhân cũng được sử dụng đánh giá bằng cách sử dụng khảo sát ưu tiên.
Hiệu quả trên người lớn và thiếu niên
Hiệu quả của điều trị dự phòng bằng HEMLIBRA có liên quan tới tỷ lệ chảy máu được điều trị được thể hiện trong Bảng 1.
- xem Bảng 1
 • HAVEN 3
• HAVEN 3
Hiệu quả của dự phòng bằng HEMLIBRA so với không dự phòng liên quan đến tỷ lệ chảy máu được điều trị, tất cả các trường hợp chảy máu, chảy máu tự phát được điều trị, chảy máu khớp và khớp đích được điều trị được thể hiện trong Bảng 2.
- xem Bảng 2
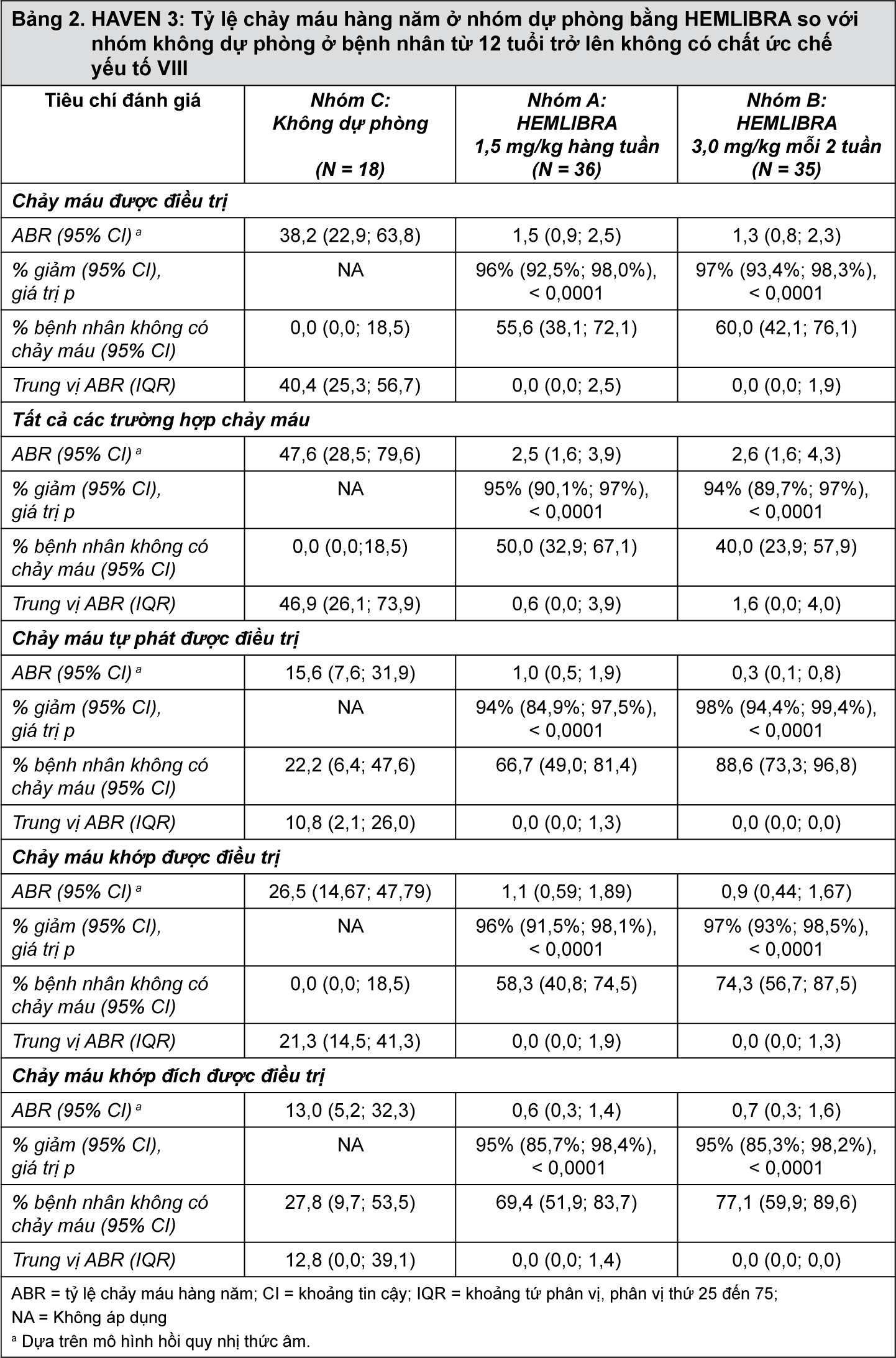
Trong phân tích trên cùng bệnh nhân thử nghiệm lâm sàng HAVEN 3, dự phòng bằng HEMLIBRA làm giảm đáng kể (p < 0,0001) (68%) tỷ lệ chảy máu đối với chảy máu được điều trị so với dự phòng bằng yếu tố VIII trước đó được tuyển chọn từ những nghiên cứu không can thiệp trước khi ghi danh.
- xem Bảng 3
 • HAVEN 1
• HAVEN 1
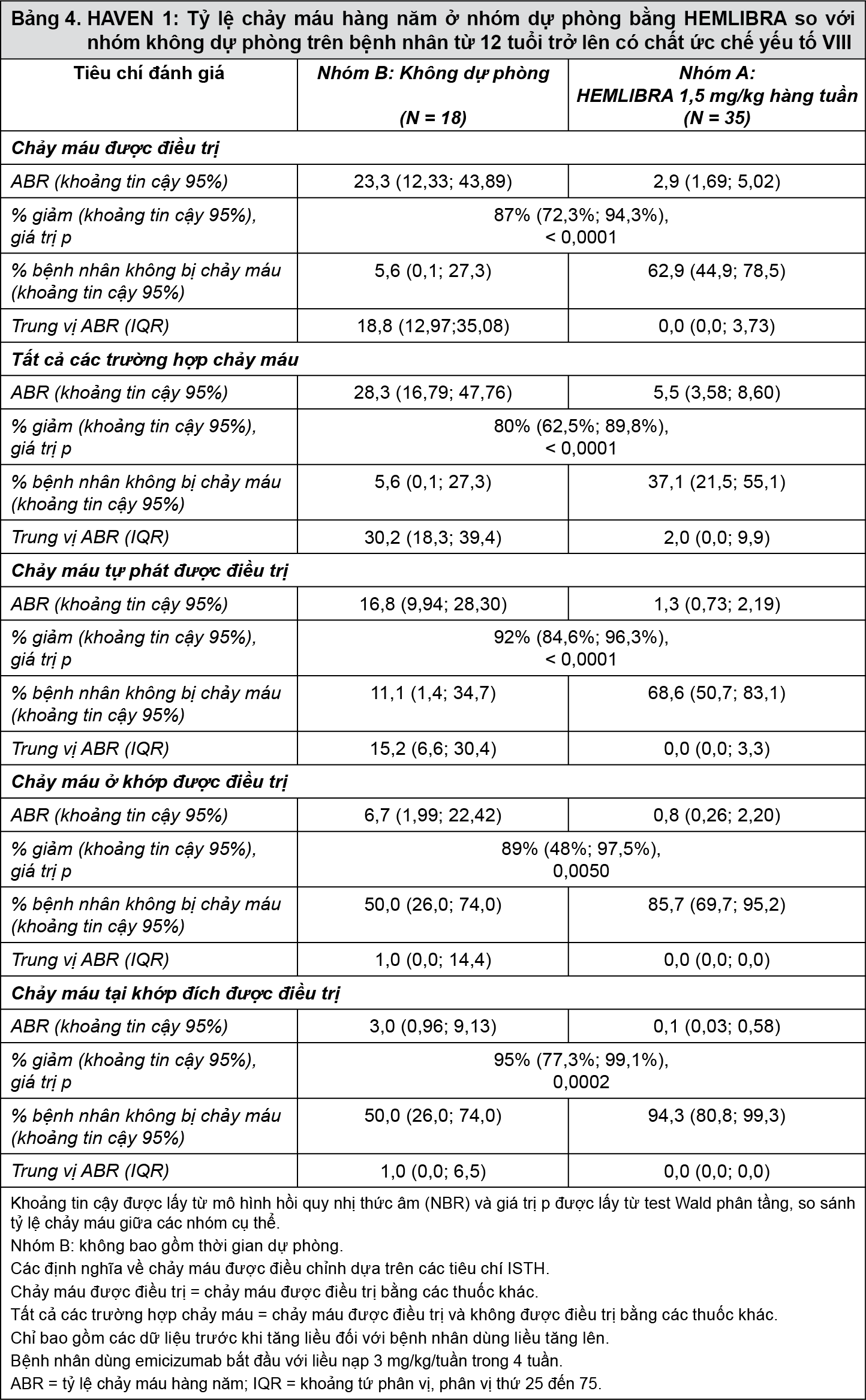
Hiệu quả của dự phòng bằng HEMLIBRA so với không dự phòng có liên quan tới tỷ lệ chảy máu được điều trị, tất cả các trường hợp chảy máu, chảy máu tự phát được điều trị, chảy máu khớp và khớp đích được điều trị được thể hiện trong Bảng 4.
- xem Bảng 4
Các phân tích bổ sung cho HAVEN 1 để đánh giá khả năng kiểm soát lâu dài các lần chảy máu với việc dự phòng bằng HEMLIBRA được thực hiện bằng cách sử dụng khoảng thời gian điều trị 12 tuần lên tới tuần 72. Khi tỷ lệ chảy máu hàng năm đối với chảy máu được điều trị được đánh giá qua khoảng thời gian 12 tuần, giá trị trung bình tỷ lệ chảy máu hàng năm giảm theo thời gian và sự cải thiện được duy trì tới tuần 72, trong khi trung vị vẫn giữ nguyên ở mức 0 (xem Bảng 5). Những dữ liệu này cho thấy hiệu quả lâu dài của dự phòng bằng HEMLIBRA. Giá trị trung bình và trung vị của tỷ lệ chảy máu hàng năm được tính toán được thể hiện trong Bảng 5.
- xem Bảng 5

Trong phân tích trên cùng bệnh nhân thử nghiệm lâm sàng HAVEN 1, dự phòng bằng HEMLIBRA làm giảm đáng kể (p = 0,0003) (79%) tỷ lệ chảy máu đối với chảy máu được điều trị so với dự phòng bằng các thuốc khác trước đó được tuyển chọn từ những nghiên cứu không can thiệp trước khi ghi danh.
- xem Bảng 6
 • HAVEN 4
• HAVEN 4
Kết quả phân tích tạm thời hiệu quả cho nghiên cứu lâm sàng HAVEN 4 được tóm tắt dưới đây. 41 bệnh nhân từ 12 tuổi trở lên được đánh giá hiệu quả với trung vị thời gian theo dõi là 17,3 tuần (trong khoảng 15,9 đến 21,1 tuần). Hiệu quả của dự phòng bằng HEMLIBRA mỗi 4 tuần liên quan tới tỷ lệ chảy máu được điều trị, tất cả các trường hợp chảy máu, chảy máu tự phát được điều trị, chảy máu tại khớp và tại khớp đích được điều trị được thể hiện trong Bảng 7.
- xem Bảng 7
 Thước đo kết cục liên quan đến sức khỏe ở người trưởng thành và thiếu niên
Thước đo kết cục liên quan đến sức khỏe ở người trưởng thành và thiếu niên
Các nghiên cứu lâm sàng trên người trưởng thành và thiếu niên được đánh giá các kết cục của bệnh nhân được báo cáo với một vài thước đo. Bảng câu hỏi về chất lượng cuộc sống của bệnh nhân người lớn mắc Haemophilia (Haem-A-QoL) và bảng câu hỏi dành cho thiếu niên (Haemo-QoL-SF, từ 8 đến dưới 18 tuổi) đánh giá chất lượng cuộc sống liên quan đến chứng hemophilia ở bệnh nhân. Đối với Haem-A-QoL và Haemo-QoL-SF, Điểm sức khỏe thể chất (ví dụ như, sưng đau, có đau khớp, đau khi di chuyển, khó khan khi đi bộ xa và cần thời gian để sẵn sàng) và tổng số điểm (tổng của tất cả các điểm) được định nghĩa giao thức là kết cục được quan tâm. Để đo lường sự thay đổi tình trạng sức khỏe, điểm chỉ số tiện ích (IUS) và thang đánh giá trực quan (VAS) từ bảng câu hỏi EuroQoL Five-Dimension 5 cấp độ (EQ-5D-5L) cũng được đưa vào kiểm tra.
Khảo sát ưu tiên Emicizumab (EmiPref), một đánh giá về ưu tiên của bệnh nhân khi điều trị, đã được sử dụng trong HAVEN 3 và 4.
Các kết cục của HAVEN 3 liên quan đến sức khỏe
Trong HAVEN 3, chất lượng cuộc sống liên quan đến sức khỏe (HRQoL) của bệnh nhân ≥ 18 tuổi được đánh giá vào tuần 25 dựa trên bộ câu hỏi chất lượng cuộc sống đặc biệt cho bệnh hemophilia (Haem-A-QoL) dành cho người lớn. Bộ câu hỏi Haem-A-QoL là biện pháp đánh giá có giá trị và đáng tin cậy về chất lượng cuộc sống của bệnh nhân liên quan đến sức khỏe.
- xem Bảng 8
 Kết quả về tình trạng sức khỏe của bệnh nhân trong HAVEN 1
Kết quả về tình trạng sức khỏe của bệnh nhân trong HAVEN 1
Trong HAVEN 1, chất lượng cuộc sống liên quan đến sức khỏe (HRQoL) của bệnh nhân ≥ 18 tuổi được đánh giá vào tuần 25 dựa trên bộ câu hỏi chất lượng cuộc sống đặc biệt cho bệnh hemophilia (Haem-A-QoL) dành cho người lớn.
- xem Bảng 9
 Kết quả HAVEN 1 về tình trạng sức khỏe
Kết quả HAVEN 1 về tình trạng sức khỏe
Trong HAVEN 1, tình trạng sức khỏe của bệnh nhân được đánh giá dựa trên bộ câu hỏi EuroQoL Five-Dimension-Five Levels (EQ-5D-5L). EQ-5D-5L là bộ câu hỏi có giá trị và đáng tin cậy về tình trạng sức khỏe.
- xem Bảng 10
 Ưu tiên bệnh nhân trong HAVEN 3 và 4
Ưu tiên bệnh nhân trong HAVEN 3 và 4
Trong HAVEN 3 và 4, bệnh nhân dùng HEMLIBRA (1 lần/tuần, mỗi 2 tuần hoặc mỗi 4 tuần) báo cáo họ có ưu tiên dùng HEMLIBRA tiêm dưới da, khi mà trước đó họ đã điều trị tiêm truyền tĩnh mạch hoặc không có ưu tiên nào ở tuần 17. Trong số những bệnh nhân trong HAVEN 3 đã trả lời bảng câu hỏi ưu tiên, 89 trong số 95 bệnh nhân (93,7%) báo cáo ưu tiên dùng HEMLIBRA thay cho điều trị tiêm truyền tĩnh mạch trước đó, và đặc biệt 45 trong số 46 bệnh nhân (97,8%) ưu tiên dùng HEMLIBRA thay cho điều trị dự phòng trước đó bằng yếu tố VIII. Trong HAVEN 4, tất cả 38 bệnh nhân (100%) trả lời bảng câu hỏi ưu tiên báo cáo ưu tiên dùng HEMLIBRA thay cho điều trị tiêm truyền tĩnh mạch trước đó.
Trong HAVEN 3 và 4, hai lý do thường được bệnh nhân xếp hạng là quan trọng nhất cho ưu tiên lựa chọn HEMLIBRA là đường dùng dễ hơn và tần suất điều trị nhỏ hơn.
Nghiên cứu lâm sàng trên bệnh nhân trẻ em
• HAVEN 2 (Phân tích tạm thời)
Điều trị dự phòng bằng HEMLIBRA hàng tuần được đánh giá trong nghiên cứu lâm sàng nhóm đơn, đa trung tâm, nhãn mở trên bệnh nhi (dưới 12 tuổi, hoặc từ 12 đến 17 tuổi cân nặng < 40 kg) bị bệnh hemophilia A có các chất ức chế yếu tố VIII. Bệnh nhân điều trị dự phòng bằng HEMLIBRA với liều 3 mg/kg hàng tuần trong vòng 4 tuần đầu, sau đó 1,5 mg/kg mỗi tuần.
Nghiên cứu đánh giá về dược động học, độ an toàn và hiệu quả bao gồm cả hiệu quả điều trị dự phòng hàng tuần bằng HEMLIBRA so với điều trị dự phòng theo đợt trước đó và điều trị dự phòng bằng các thuốc khác trên bệnh nhân tham gia nghiên cứu không can thiệp trước khi tham gia nghiên cứu này (so sánh trên cùng bệnh nhân).
Kết quả đánh giá hiệu quả trong HAVEN 2 (Phân tích tạm thời)
Tại thời điểm đánh giá tạm thời, hiệu quả được đánh giá trên 59 bệnh nhân nhi dưới 12 tuổi đã dùng HEMLIBRA dự phòng hàng tuần trong ít nhất 12 tuần, bao gồm 38 bệnh nhân tuổi từ 6 đến dưới 12; 17 bệnh nhân tuổi từ 2 đến dưới 6 và 4 bệnh nhân dưới 2 tuổi. Tỷ lệ chảy máu hàng năm và tỷ lệ phần trăm bệnh nhân không bị chảy máu được tính toán trên 59 bệnh (xem Bảng 11). Trung vị thời gian theo dõi các bệnh nhân này là 29,6 tuần (dao động từ 18,4 đến 63).
- xem Bảng 11

Trong phân tích tạm trên cùng bệnh nhân, điều trị dự phòng bằng HEMLIBRA hàng tuần làm giảm có ý nghĩa lâm sàng (98%) tỷ lệ chảy máu trong các trường hợp chảy máu được điều trị trên 18 bệnh nhi đã dự phòng bằng HEMLIBRA trong ít nhất 12 tuần so với tỷ lệ chảy máu ghi nhận được trong nghiên cứu không can thiệp trước đó.
- xem Bảng 12
 Đánh giá kết cục liên quan đến sức khỏe trên bệnh nhân nhi
Đánh giá kết cục liên quan đến sức khỏe trên bệnh nhân nhi
Kết cục liên quan đến sức khỏe trong HAVEN 2
Trong HAVEN 2, chất lượng cuộc sống của bệnh nhân từ 8 tuổi đến dưới 12 tuổi được đánh giá ở tuần thứ 25 dựa trên bảng câu hỏi Haemo-QoL-SF dành cho trẻ em. Bộ câu hỏi Haemo-QoL-SF là biện pháp đánh giá chất lượng cuộc sống có giá trị và đáng tin cậy.
- xem Bảng 13

Trong HAVEN 2, chất lượng cuộc sống của bệnh nhân dưới 12 tuổi cũng được đánh giá ở tuần 25 dựa trên bộ câu hỏi được điều chỉnh InhibQoL trên phương diện khó khăn về phía người chăm sóc được hoàn thành bởi người chăm sóc. Bộ câu hỏi được điều chỉnh InhibQoL là biện pháp đánh giá có giá trị và đáng tin cậy về chất lượng cuộc sống của bệnh nhân.
- xem Bảng 14
 Phẫu thuật và các thủ thuật y tế trong các thử nghiệm lâm sàng HAVEN
Phẫu thuật và các thủ thuật y tế trong các thử nghiệm lâm sàng HAVEN
Có rất ít kinh nghiệm trên những bệnh nhân sử dụng các thuốc khác hoặc yếu tố VIII trong các phẫu thuật hoặc các thủ thuật y tế ở những bệnh nhân dự phòng bằng HEMLIBRA. Các thuốc khác hoặc yếu tố VIII sử dụng trong các phẫu thuật và các thủ thuật y tế được xác định bởi điều tra viên.
Tính sinh miễn dịch
Tương tự các protein được sử dụng trong điều trị, HEMLIBRA có khả năng gây đáp ứng miễn dịch cho bệnh nhân. Không có bệnh nhân nào được xét nghiệm dương tính với kháng thể kháng emicizumab trong các thử nghiệm lâm sàng HAVEN 1, HAVEN 2, HAVEN 3 và HAVEN 4. 4 bệnh nhân được xét nghiệm dương tính với kháng thể kháng emicizumab trong nghiên cứu lâm sàng pha I/II (N=18), tất cả trong số này đều thuộc loại không có tính chất trung hòa kháng nguyên.
Dữ liệu này phản ánh số lượng bệnh nhân có kết quả xét nghiệm dương tính với kháng thể kháng emicizumab bằng xét nghiệm ELISA. Kết quả thử nghiệm khả năng sinh miễn dịch có thể bị ảnh hưởng bởi nhiều yếu tố bao gồm thử nghiệm độ nhạy và độ đặc hiệu, các thao tác xử lý mẫu, thời điểm thu mẫu, các thuốc dùng đồng thời và bệnh lý kèm theo. Vì những lý do này, việc so sánh tỷ lệ xuất hiện kháng thể kháng emicizumab với tỷ lệ xuất hiện kháng thể kháng các thuốc khác có thể bị nhầm lẫn.
ĐẶC TÍNH DƯỢC ĐỘNG HỌC
Đặc tính dược động học của emicizumab được xác định qua phân tích không ngăn (noncompartmental analysis) trên người tình nguyện khỏe mạnh và sử dụng phân tích dược động học quần thể dựa trên cơ sở dữ liệu 389 bệnh nhân bị bệnh hemophilia A.
Hấp thu
Sau khi tiêm dưới da cho bệnh nhân bị bệnh hemophilia A, thời gian bán hấp thu của thuốc là 1,6 ngày.
Sau khi tiêm dưới da liều lặp lại 3 mg/kg 1 lần/tuần trong 4 tuần đầu trên bệnh nhân bị bệnh hemophilia A, nồng độ đáy trung bình trong huyết tương của emicizumab đạt 52,6±13,6 µg/mL ở tuần thứ 5. Nồng độ đáy trung bình kéo dài trong huyết tương của emicizumab ở trạng thái ổn định là 51,1 µg/mL, 46,7 µg/mL và 38,3 µg/mL với liều duy trì được khuyến cáo tương ứng là 1,5 mg/kg 1 lần/tuần, 3 mg/kg mỗi 2 tuần hoặc 6 mg/kg mỗi 4 tuần (Hình 1, Bảng 15).
- xem Hình 1
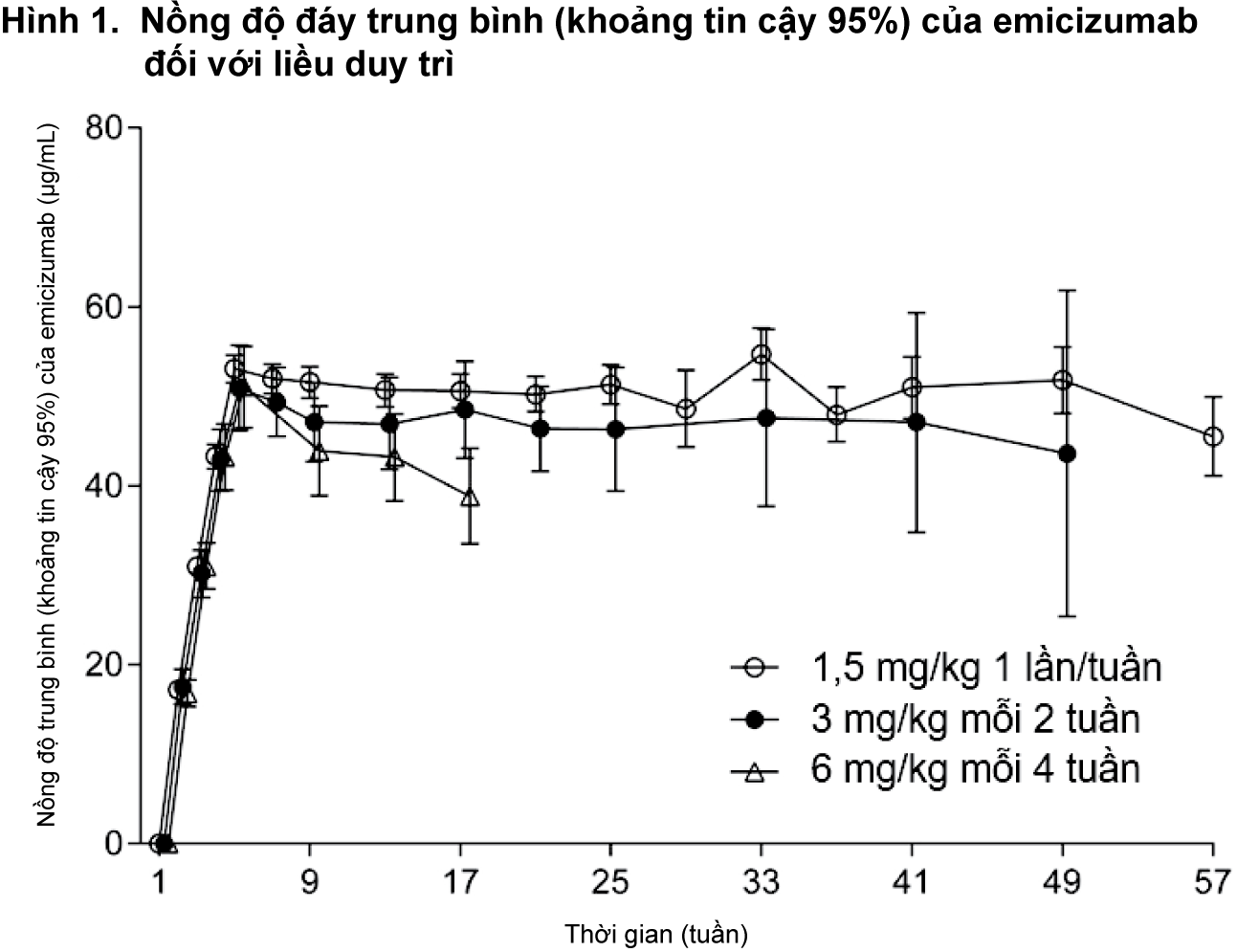
Giá trị trung bình (±SD) của nồng độ đáy (C
trough), nồng độ đỉnh (C
max) và tỷ lệ trung bình C
max/C
trough đối với liều duy trì được khuyến cáo là 1,5 mg/kg 1 lần/tuần, 3 mg/kg mỗi 2 tuần hoặc 6 mg/kg mỗi 4 tuần được thể hiện trong Bảng 15.
- xem Bảng 15

Dữ liệu dược động học tương tự được tiếp tục theo dõi sau liều dùng hàng tuần (3 mg/kg/tuần cho mỗi 4 tuần sau khi dùng liều 1,5 mg/kg/tuần) ở người lớn/thiếu niên (từ 12 tuổi trở lên) và trẻ em (dưới 12 tuổi).
- xem Hình 2
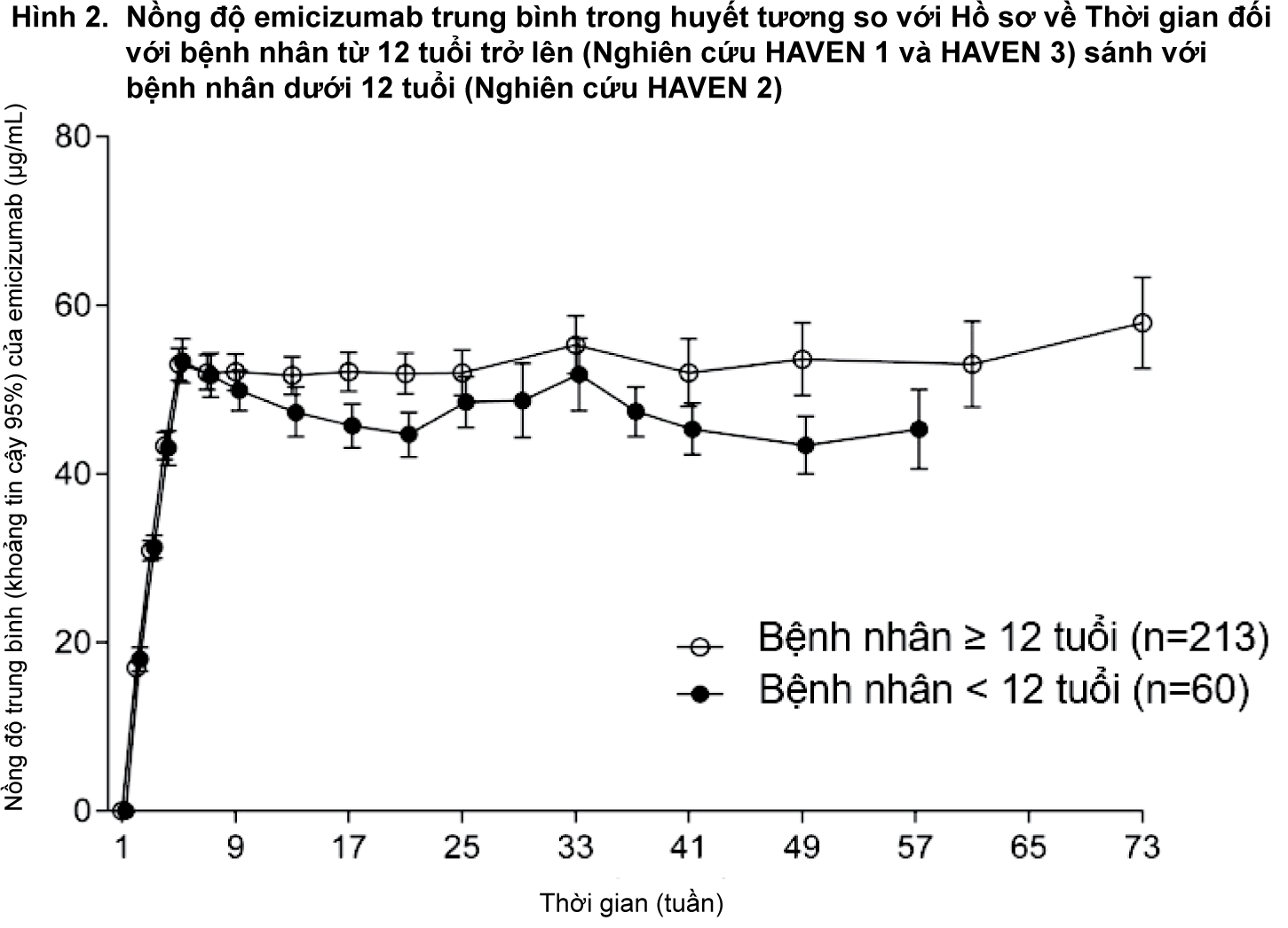
Trên người tình nguyện khỏe mạnh, sinh khả dụng tuyệt đối sau khi tiêm dưới da liều 1 mg/kg nằm trong khoảng 80,4% và 93,1% phụ thuộc vị trí tiêm. Dữ liệu dược động học tương tự cũng được ghi nhận sau khi tiêm dưới da vùng bụng, phía trên cánh tay và đùi. Emicizumab có thể được tiêm thay đổi tại các vị trí này (xem mục
Liều lượng và cách dùng).
Phân bố
Sau khi tiêm tĩnh mạch liều đơn 0,25 mg/kg emicizumab trên người tình nguyện khỏe mạnh, thể tích phân bố ở trạng thái ổn định là 106 mL/kg (ví dụ 7,4 L cho 1 người lớn cân nặng 70). Không dùng emicizumab đường tĩnh mạch (xem mục
Liều lượng và cách dùng).
Thể tích phân bố biểu kiến (V/F) của emicizumab, ước tính từ phân tích dược động học quần thể trên bệnh nhân bị bệnh hemophilia A sau khi tiêm dưới da liều lặp lại là 10,4 L.
Chuyển hóa
Quá trình chuyển hóa của emicizumab chưa được nghiên cứu. Kháng thể IgG chủ yếu được chuyển hóa qua quá trình ly giải protein tại lysosome, sau đó được cơ thể thải trừ hoặc tái sử dụng.
Thải trừ
Sau khi tiêm tĩnh mạch liều 0,25 mg/kg trên người tình nguyện khỏe mạnh, thanh thải toàn phần của emicizumab là 3,26 mL/kg/ngày (ví dụ 0,228 L/ngày đối với 1 người lớn cân nặng 70 kg) và thời gian bán thải trung bình pha cuối là 26,7 ngày.
Sau khi tiêm dưới da liều đơn cho người tình nguyện khỏe mạnh, thời gian bán thải thải trừ khoảng 4 đến 5 tuần.
Sau khi tiêm dưới da liều lặp lại cho bệnh nhân bị bệnh hemophilia A, thanh thải khả kiến là 0,272 L/ngày và thời gian bán thải thải trừ khả kiến là 26,8 ngày.
Tuyến tính theo liều
Các thông số dược động học của emicizumab tỷ lệ với liều trên bệnh nhân bị bệnh hemophilia A trong khoảng liều dao động từ 0,3 đến 6 mg/kg, 1 lần/ tuần sau khi tiêm dưới da.
Dược động học trên các quần thể đặc biệt
Suy giảm chức năng thận
Chưa có nghiên cứu chuyên biệt về ảnh hưởng của suy giảm chức năng thận đến dược động học của emicizumab được thực hiện. Hầu hết bệnh nhân mắc hemophilia A trong quần thể phân tích dược động học có chức năng thận bình thường (N=332; độ thanh thải creatinine [CLcr] ≥ 90 mL/phút hoặc suy thận nhẹ (N=27; độ thanh thải creatinine 60-89 mL/phút). Chỉ có 2 bệnh nhân có suy thận trung bình (độ thanh thải creatinine 30-59 mL/phút). Không có bệnh nhân bị suy giảm chức năng thận ở mức độ nặng trong các nghiên cứu lâm sàng. Suy thận nhẹ và trung bình dường như không ảnh hưởng đến dược động học của emicizumab. Không cần điều chỉnh liều cho bệnh nhân suy thận.
Suy giảm chức năng gan
Chưa có nghiên cứu chuyên biệt về ảnh hưởng của suy giảm chức năng gan đến dược động học của emicizumab được thực hiện. Phần lớn bệnh nhân bị bệnh hemophilia A trong phân tích dược động học quần thể có chức năng gan bình thường (bilirubin và AST ≤ giới hạn trên của mức bình thường, N=300) hoặc suy giảm nhẹ chức năng gan (bilirubin ≤ giới hạn trên của mức bình thường và AST > giới hạn trên của mức bình thường hoặc bilirubin < 1,0 đến 1,5 lần giới hạn trên của mức bình thường và bất cứ giá trị AST nào, N=51). Chỉ có 6 bệnh nhân có suy gan trung bình (1,5 lần giới hạn trên của mức bình thường < bilirubin ≤ 3 lần giới hạn trên của mức bình thường và AST bất kỳ). Suy giảm chức năng gan ở mức độ nhẹ hoặc trung bình không ảnh hưởng đến dược động học của emicizumab (xem thêm mục
Liều lượng và cách dùng - Các hướng dẫn đặc biệt về liều). Suy giảm chức năng gan được định nghĩa theo tiêu chuẩn của viện ung thư quốc gia (NCI) về rối loạn chức năng gan.
Bệnh nhi
Ảnh hưởng của tuổi đến dược động học của emicizumab được đánh giá qua phân tích dược động học quần thể bao gồm 5 trẻ nhũ nhi (từ 1 tháng đến dưới 2 tuổi), 55 trẻ em (từ 2 tuổi đến <12 tuổi) và 50 trẻ vị thành niên (12 đến < 18 tuổi) bị bệnh hemophilia A. Tuổi không ảnh hưởng đến dược động học của emicizumab trên bệnh nhi (xem mục
Liều lượng và cách dùng - Các hướng dẫn đặc biệt về liều).
Bệnh nhân cao tuổi
Ảnh hưởng của tuổi đến dược động học của emicizumab được đánh giá dựa trên phân tích dược động học quần thể bao gồm 13 bệnh nhân từ 65 tuổi trở lên (không có bệnh nhân nào trên 75 tuổi). Sinh khả dụng của thuốc tăng lên theo tuổi, nhưng không có sự khác biệt có ý nghĩa lâm sàng được ghi nhận liên quan đến dược động học của emicizumab giữa bệnh nhân < 65 tuổi và bệnh nhân ≥ 65 tuổi.
Chủng tộc
Các phân tích dược động học quần thể trên bệnh nhân bị bệnh hemophilia A cho thấy chủng tộc không ảnh hưởng đến dược động học của emicizumab. Không cần hiệu chỉnh liều theo yếu tố chủng tộc.
AN TOÀN TIỀN LÂM SÀNG
Các dữ liệu tiền lâm sàng cho thấy thuốc không gây nguy hại đặc biệt nào trên người dựa trên nghiên cứu độc tính cấp và nghiên cứu liều lặp lại, bao gồm cả các tiêu chí đánh giá về độ an toàn dược lý và độc tính trên sinh sản.
Khả năng sinh ung thư
Chưa có các nghiên cứu về khả năng sinh ung thư được thực hiện để xác định khả năng sinh ung thư của emicizumab.
Độc tính trên gen
Chưa có các nghiên cứu được thực hiện để xác định khả năng gây đột biến gen của emicizumab.
Suy giảm khả năng sinh sản
Emicizumab không gây ra bất cứ thay đổi độc tính nào trên cơ quan sinh sản của khỉ cynomolgus đực và cái ở mức liều lên đến 30 mg/kg/tuần trong các nghiên cứu độc tính toàn thân khi tiêm dưới da trong thời gian lên đến 26 tuần và ở mức liều lên đến 100 mg/kg/tuần trong nghiên cứu độc tính toàn thân đường tiêm tĩnh mạch trong 4 tuần.
Độc tính sinh sản
Hiện chưa có các dữ liệu về tác dụng phụ có khả năng xảy ra của emicizumab trên sự phát triển của phôi - thai.
Các vấn đề khác
Trong 1 nghiên cứu
in vitro về giải phóng các cytokine sử dụng máu của người tình nguyện khỏe mạnh, nồng độ các cytokins sinh ra bởi emicizumab tương đương với các kháng thể nguy cơ thấp khác.






 Đăng xuất
Đăng xuất
