Pharmacotherapeutic group: Hormone antagonists and related agents, anti-androgens.
ATC code: L02BB04.
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Prostate cancer is known to be androgen sensitive and responds to inhibition of androgen receptor signalling. Despite low or even undetectable levels of serum androgen, androgen receptor signalling continues to promote disease progression. Stimulation of tumour cell growth via the androgen receptor requires nuclear localization and DNA binding. Enzalutamide is a potent androgen receptor signalling inhibitor that blocks several steps in the androgen receptor signalling pathway. Enzalutamide competitively inhibits androgen binding to androgen receptors, and consequently; inhibits nuclear translocation of activated receptors and inhibits the association of the activated androgen receptor with DNA even in the setting of androgen receptor overexpression and in prostate cancer cells resistant to anti-androgens. Enzalutamide treatment decreases the growth of prostate cancer cells and can induce cancer cell death and tumour regression. In preclinical studies enzalutamide lacks androgen receptor agonist activity.
Pharmacodynamic effects: In a phase 3 clinical trial (AFFIRM) of patients who failed prior chemotherapy with docetaxel, 54% of patients treated with enzalutamide, versus 1.5% of patients who received placebo, had at least a 50% decline from baseline in PSA levels.
In another phase 3 clinical trial (PREVAIL) in chemo-naïve patients, patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 78.0% versus 3.5% (difference = 74.5%, p < 0.0001).
In a phase 2 clinical trial (TERRAIN) in chemo-naïve patients, patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving bicalutamide, 82.1% versus 20.9% (difference = 61.2%, p < 0.0001).
In the MDV3100-09 clinical trial (STRIVE) of non-metastatic and metastatic CRPC, patients receiving enzalutamide demonstrated a significantly higher total confirmed PSA response rate (defined as a ≥ 50% reduction from baseline) compared with patients receiving bicalutamide, 81.3% versus 31.3% (difference = 50.0%, p < 0.0001).
In the MDV3100-14 clinical trial (PROSPER) of non-metastatic CRPC, patients receiving enzalutamide demonstrated a significantly higher confirmed PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 76.3% versus 2.4% (difference = 73.9%, p < 0.0001).
Clinical efficacy and safety: Efficacy of enzalutamide was established in three randomized placebo-controlled multicentre phase 3 clinical studies [MDV3100-14 (PROSPER), CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] of patients with progressive metastatic prostate cancer who had disease progression on androgen deprivation therapy [luteinising hormone-releasing hormone (LHRH) analogue or after bilateral orchiectomy]. The PREVAIL study enrolled metastatic CRPC chemotherapy-naïve patients; whereas the AFFIRM study enrolled metastatic CRPC patients who had received prior docetaxel; and the PROSPER study enrolled patients with non-metastatic CRPC. Additionally, efficacy in patients with mHSPC was also established in one randomized, placebo-controlled multicentre phase 3 clinical study [9785-CL-0335 (ARCHES)]. All patients continued on a LHRH analogue or had bilateral orchiectomy.
In the active treatment arm, Xtandi was administered orally at a dose of 160 mg daily. In the four clinical studies (ARCHES, PROSPER, AFFIRM and PREVAIL), patients received placebo in the control arm and patients were allowed, but not required, to take prednisone (maximum daily dose allowed was 10 mg prednisone or equivalent).
Changes in PSA serum concentration independently do not always predict clinical benefit. Therefore, in the four studies it was recommended that patients be maintained on their study treatments until discontinuation criteria were met as specified as follows for each study.
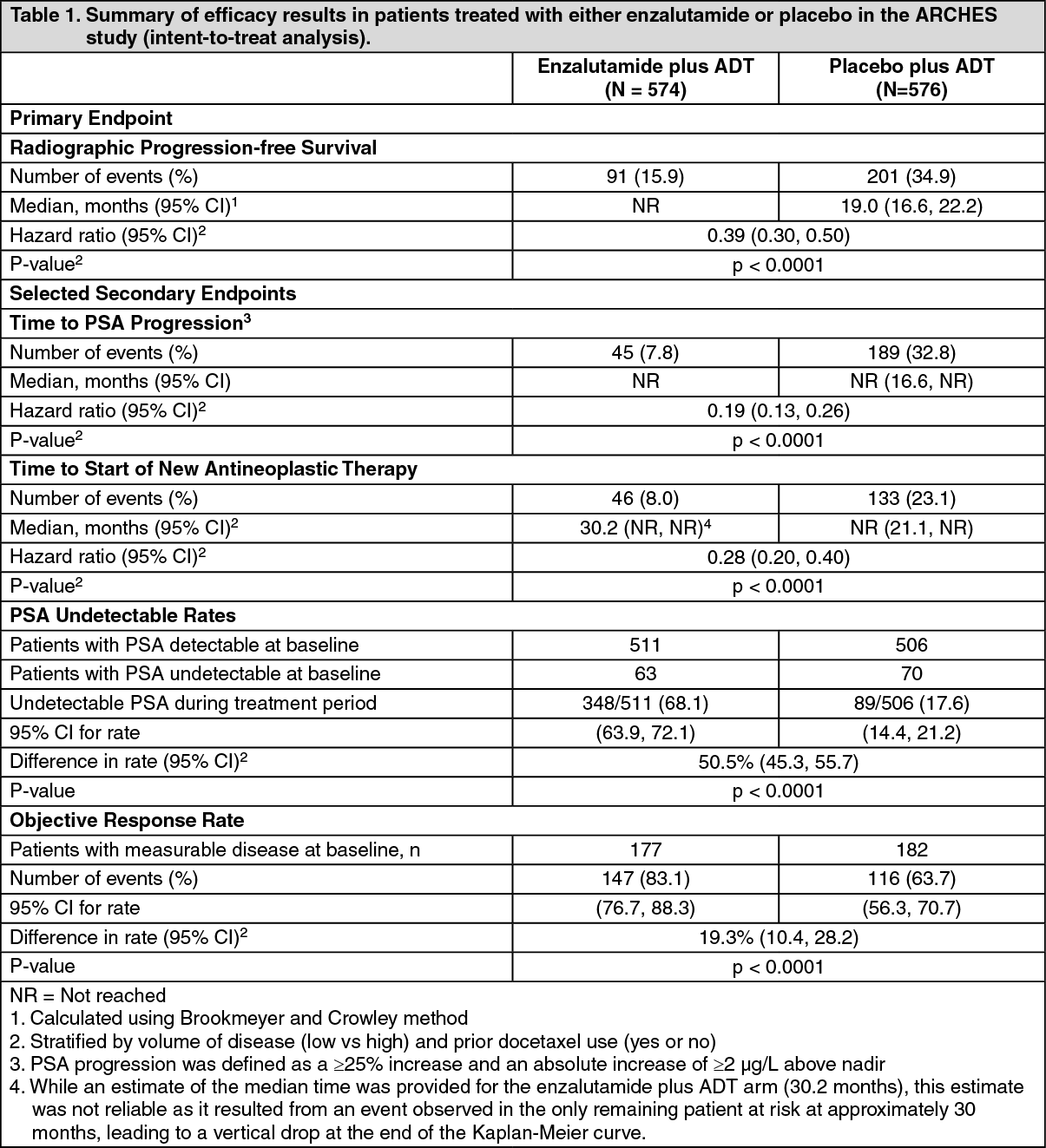
9785-CL-0335 (ARCHES) Study (patients with metastatic HSPC): The ARCHES study enrolled 1150 patients with mHSPC randomized 1:1 to receive treatment with enzalutamide plus ADT or placebo plus ADT (ADT defined as LHRH analogue or bilateral orchiectomy). Patients received enzalutamide at 160 mg once daily (N=574) or placebo (N=576). The demographic and baseline characteristics were well balanced between the two treatment groups. The median age at randomization was 70 years in both treatment groups. Most patients in the total population were Caucasian (80.5%); 13.5% were Asian and 1.4% were Black. The Eastern Cooperative Oncology Group Performance Status (ECOG PS) score was 0 for 78% of patients and 1 for 22% of patients at study entry.
Radiographic progression-free survival (rPFS), based on independent central review, was the primary endpoint defined as the time from randomization to the first objective evidence of radiographic disease progression or death (due to any cause from time of randomization up until 24 weeks from study drug discontinuation), whichever occurred first. Key secondary efficacy endpoints assessed in the study were time to PSA progression, time to start of new antineoplastic therapy, PSA undetectable rate (decline to <0.2 μg/L), objective response rate (RECIST 1.1) based on independent review, and overall survival. See Table 1 as follows.
Enzalutamide demonstrated a statistically significant 61% reduction in the risk of an rPFS event compared to placebo [HR = 0.39 (95% CI: 0.30, 0.50); p < 0.0001]. The median time to an rPFS event was not reached in the enzalutamide arm and was 19.0 months (95% CI: 16.6, 22.2) in the placebo arm. (See Table 1 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Enzalutamide demonstrated a statistically significant 81.0% reduction in the risk of PSA progression compared with placebo [HR = 0.19 (95% CI: 0.13, 0.26); p < 0.0001]. The median time to PSA progression (95% CI) was not reached for enzalutamide or placebo.
Enzalutamide demonstrated a statistically significant 72% reduction in risk of initiation of a new antineoplastic therapy compared to placebo [HR=0.28 (95% CI: 0.20, 0.40); p<0.0001].
Enzalutamide significantly increased the rate of a PSA decline to an undetectable level (< 0.2 μg/L) compared to treatment with placebo. The PSA undetectable rate was 68.1% for enzalutamide and 17.6% for placebo. The rate difference is statistically significant [50.5% (95% CI: 45.3, 55.7); p<0.0001].
The objective response rate (calculated as percentage of patients with measurable disease at baseline who achieved a complete or partial response in their soft tissue disease) was 83.1% for patients in the enzalutamide treatment arm and 63.7% in the placebo arm. Enzalutamide demonstrated a statistically significant 19.3% improvement in objective response rate compared to placebo.
The first pre-specified interim analysis for overall survival was conducted at the time of the rPFS analysis. At the time of the first interim analysis, overall survival data were not mature and did not show a statistically significant difference in patients treated with enzalutamide compared to placebo [HR = 0.81 (95% CI: 0.53, 1.25), p = 0.3361]. The median overall survival was not reached in either treatment group.
ANZUP 1304 (ENZAMET) Study (patients with metastatic HSPC): The ENZAMET study enrolled 1125 patients with mHSPC randomized 1:1 to receive treatment orally once daily with enzalutamide 160 mg (N=563) or nonsteroidal anti-androgen (NSAA, N=562). All patients in the trial received an LHRH analog or had a prior bilateral orchiectomy. Patients were stratified by volume of disease (low vs high), concomitant antiresorptive therapy (yes vs no), comorbidities (ACE-27: 0 to 1 vs 2 to 3) and planned use of a total of 6 cycles of docetaxel, of which 0-2 cycles were allowed before randomization (yes vs no). Patients were required to have confirmation of metastatic prostate cancer by positive bone scan or metastatic lesions on CT or MRI scan. Patients continued treatment until evidence of clinical progression via CT, MRI or whole body bone scan.
The following patient demographics and baseline characteristics were balanced between the two treatment arms. The median age at randomization was 69 years in the enzalutamide group and 68 years in the NSAA group (treated with bicalutamide, nilutamide, or flutamide). The majority of patients had an ECOG performance status score of 0 (72%) and a Gleason score of ≥ 8 (58%). Forty-eight percent (48%) of patients had a low volume of disease and 52% of patients had a high volume of disease. High volume of disease is defined as metastases involving the viscera or, in the absence of visceral lesions, there must be 4 or more bone lesions, at least 1 of which must be in a bony structure beyond the vertebral column and pelvic bone. Ten percent (10%) of patients had concomitant antiresorptive therapy; 75% had no or mild comorbidities (ACE-27 score of 0 to 1) and 45% had a total of 6 cycles of docetaxel, of which 0-2 cycles were allowed before randomization.
At the time of analysis, median follow-up for OS was 33.8 months. The interim analysis demonstrated a statistically significant 33% reduction in the risk of death for patients treated with enzalutamide compared to conventional NSAA treatment [HR of 0.67 (95% CI: 0.52, 0.86; p=0.0018)]. (See Figures 3 and 4).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
MDV3100-14 (PROSPER) study (patients with non-metastatic CRPC): The PROSPER study enrolled 1401 patients with asymptomatic, high-risk non-metastatic CRPC who continued on androgen deprivation therapy (ADT; defined as LHRH analogue or prior bilateral orchiectomy). Patients were required to have a PSA doubling time ≤ 10 months, PSA ≥ 2 ng/mL, and confirmation of non-metastatic disease by blinded independent central review (BICR).
Patients with a history of mild to moderate heart failure (NYHA Class I or II), and patients taking medicinal products associated with lowering the seizure threshold were allowed. Patients were excluded with a previous history of seizure, a condition that might predispose them to seizure, or certain prior treatments for prostate cancer (i.e., chemotherapy, ketoconazole, abiraterone acetate, aminoglutethimide and/or enzalutamide).
Patients were randomized 2:1 to receive either enzalutamide at a dose of 160 mg once daily (N = 933) or placebo (N = 468). Patients were stratified by Prostate Specific Antigen (PSA) Doubling Time (PSADT) (< 6 months or ≥ 6 months) and the use of bone-targeting agents (yes or no).
The demographic and baseline characteristics were well-balanced between the two treatment arms. The median age at randomization was 74 years in the enzalutamide arm and 73 years in the placebo arm. Most patients (approximately 71%) in the study were Caucasian; 16% were Asian and 2% were Black. Eighty-one percent (81%) of patients had an ECOG performance status score of 0 and 19% patients had an ECOG performance status of 1.
Metastasis-free survival (MFS) was the primary endpoint defined as the time from randomization to radiographic progression or death within 112 days of treatment discontinuation without evidence of radiographic progression, whichever occurred first. Key secondary endpoints assessed in the study were time to PSA progression, time to first use of new antineoplastic therapy (TTA), overall survival (OS). Additional secondary endpoints included time to first use of cytotoxic chemotherapy and chemotherapy-free survival. See results as follows (Table 2).
Enzalutamide demonstrated a statistically significant 71% reduction in the relative risk of radiographic progression or death compared to placebo [HR = 0.29 (95% CI: 0.24, 0.35), p < 0.0001]. Median MFS was 36.6 months (95% CI: 33.1, NR) on the enzalutamide arm versus 14.7 months (95% CI: 14.2, 15.0) on the placebo arm. Consistent MFS results were also observed in all pre-specified patient subgroups including PSADT (< 6 months or ≥ 6 months), demographic region (North America, Europe, rest of world), age (< 75 or ≥ 75), use of a prior bone-targeting agent (yes or no). (See Table 2 and Figure 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the final analysis for overall survival, conducted when 466 deaths were observed, a statistically significant improvement in overall survival was demonstrated in patients randomized to receive enzalutamide compared with patients randomized to receive placebo with a 26.6% reduction in risk of death [hazard ratio (HR) = 0.734, (95% CI: 0.608; 0.885), p = 0.0011]. The median follow-up time was 48.6 and 47.2 months for the enzalutamide and placebo groups, respectively. Thirty-three percent of enzalutamide-treated and 65% of placebo-treated patients received at least one subsequent antineoplastic therapy that may prolong overall survival. (See Figure 6.)
Click on icon to see table/diagram/image
Enzalutamide demonstrated a statistically significant 93% reduction in the relative risk of PSA progression compared to placebo [HR = 0.07 (95% CI: 0.05, 0.08), p < 0.0001]. Median time to PSA progression was 37.2 months (95% CI: 33.1, NR) on the enzalutamide arm versus 3.9 months (95% CI: 3.8, 4.0) on the placebo arm.
Enzalutamide demonstrated a statistically significant delay in the time to first use of new antineoplastic therapy compared to placebo [HR = 0.21 (95% CI: 0.17, 0.26), p < 0.0001]. Median time to first use of new antineoplastic therapy was 39.6 months (95% CI: 37.7, NR) on the enzalutamide arm versus 17.7 months (95% CI: 16.2, 19.7) on the placebo arm. (See Figure 7.)
Click on icon to see table/diagram/image
MDV3100-09 (STRIVE) study (chemotherapy-naïve patients with non-metastatic/metastatic CRPC): The STRIVE study enrolled 396 non-metastatic or metastatic CRPC patients who had serologic or radiographic disease progression despite primary androgen deprivation therapy who were randomized to receive either enzalutamide at a dose of 160 mg once daily (N = 198) or bicalutamide at a dose of 50 mg once daily (N = 198). PFS was the primary endpoint defined as the time from randomization to the earliest objective evidence of radiographic progression, PSA progression, or death on study. Median PFS was 19.4 months (95% CI: 16.5, not reached) in the enzalutamide group versus 5.7 months (95% CI: 5.6, 8.1) in the bicalutamide group [HR = 0.24 (95% CI: 0.18, 0.32), p < 0.0001]. Consistent benefit of enzalutamide over bicalutamide on PFS was observed in all pre-specified patient subgroups. For the non-metastatic subgroup (N = 139) a total of 19 out of 70 (27.1%) patients treated with enzalutamide and 49 out of 69 (71.0%) patients treated with bicalutamide had PFS events (68 total events). The hazard ratio was 0.24 (95% CI: 0.14, 0.42) and the median time to a PFS event was not reached in the enzalutamide group versus 8.6 months in the bicalutamide group. (See Figure 8.)
Click on icon to see table/diagram/image
9785-CL-0222 (TERRAIN) study (chemotherapy-naïve patients with metastatic CRPC): The TERRAIN study enrolled 375 chemo- and antiandrogen-therapy naïve patients with metastatic CRPC who were randomized to receive either enzalutamide at a dose of 160 mg once daily (N = 184) or bicalutamide at a dose of 50 mg once daily (N = 191). Median PFS was 15.7 months for patients on enzalutamide versus 5.8 months for patients on bicalutamide [HR = 0.44 (95% CI: 0.34, 0.57), p < 0.0001]. Progression-free survival was defined as objective evidence of radiographic disease progression by independent central review, skeletal-related events, initiation of new antineoplastic therapy or death by any cause, whichever occurred first. Consistent PFS benefit was observed across all pre-specified patient subgroups.
MDV3100-03 (PREVAIL) study (chemotherapy-naïve patients with metastatic CRPC): A total of 1717 asymptomatic or mildly symptomatic chemotherapy-naïve patients were randomized 1:1 to receive either enzalutamide orally at a dose of 160 mg once daily (N = 872) or placebo orally once daily (N = 845). Patients with visceral disease, patients with a history of mild to moderate heart failure (NYHA Class I or II), and patients taking medications associated with lowering the seizure threshold were allowed. Patients with a previous history of seizure or a condition that might predispose to seizure and patients with moderate or severe pain from prostate cancer were excluded. Study treatment continued until disease progression (evidence of radiographic progression, a skeletal-related event, or clinical progression) and the initiation of either a cytotoxic chemotherapy or an investigational agent, or until unacceptable toxicity.
Patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 71 years (range 42-93) and the racial distribution was 77% Caucasian, 10% Asian, 2% Black and 11% other or unknown races. Sixty-eight percent (68%) of patients had an ECOG performance status score of 0 and 32% patients had an ECOG performance status of 1. Baseline pain assessment was 0-1 (asymptomatic) in 67% of patients and 2-3 (mildly symptomatic) in 32% of patients as defined by the Brief Pain Inventory Short Form (worst pain over past 24 hours on a scale of 0 to 10). Approximately 45% of patients had measurable soft tissue disease at study entry, and 12% of patients had visceral (lung and/or liver) metastases.
Co-primary efficacy endpoints were overall survival and radiographic progression-free survival (rPFS). In addition to the co-primary endpoints, benefit was also assessed using time to initiation of cytotoxic chemotherapy, best overall soft tissue response, time to first skeletal-related event, PSA response (≥50% decrease from baseline), time to PSA progression, and time to FACT-P total score degradation.
Radiographic progression was assessed with the use of sequential imaging studies as defined by Prostate Cancer Clinical Trials Working Group 2 (PCWG2) criteria (for bone lesions) and/or Response Evaluation Criteria in Solid Tumors (RECIST v 1.1) criteria (for soft tissue lesions). Analysis of rPFS utilized centrally-reviewed radiographic assessment of progression.
At the pre-specified interim analysis for overall survival when 540 deaths were observed, treatment with enzalutamide demonstrated a statistically significant improvement in overall survival compared to treatment with placebo with a 29.4% reduction in risk of death [HR=0.71, (95% CI: 0.60; 0.84), p < 0.0001]. An updated survival analysis was conducted when 784 deaths were observed. Results from this analysis were consistent with those from the interim analysis (Table 3). At the updated analysis 52% of enzalutamide-treated and 81% of placebo-treated patients had received subsequent therapies for metastatic CRPC that may prolong overall survival.
A final analysis of 5-year PREVAIL data showed a statistically significant increase in overall survival was maintained in patients treated with enzalutamide compared to placebo [HR = 0.835, (95% CI: 0.75, 0.93); p-value = 0.0008] despite 28% of patients on placebo crossing over to enzalutamide. The 5-year OS rate was 26% for the enzalutamide arm compared to 21% for the placebo arm. (See Table 3 and Figures 9 and 10.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the pre-specified rPFS analysis, a statistically significant improvement was demonstrated between the treatment groups with an 81.4% reduction in risk of radiographic progression or death [HR = 0.19 (95% CI: 0.15, 0.23), p < 0.0001]. One hundred and eighteen (14%) enzalutamide-treated patients and 321 (40%) of placebo-treated patients had an event. The median rPFS was not reached (95% CI: 13.8, not reached) in the enzalutamide-treated group and was 3.9 months (95% CI: 3.7, 5.4) in the placebo-treated group (Figure 11). Consistent rPFS benefit was observed across all pre-specified patient subgroups (e.g., age, baseline ECOG performance, baseline PSA and LDH, Gleason score at diagnosis, and visceral disease at screening). A pre-specified follow-up rPFS analysis based on the investigator assessment of radiographic progression demonstrated a statistically significant improvement between the treatment groups with a 69.3% reduction in risk of radiographic progression or death [HR = 0.31 (95% CI: 0.27, 0.35), p < 0.0001]. The median rPFS was 19.7 months in the enzalutamide group and 5.4 months in the placebo group. (See Figure 11.)
Click on icon to see table/diagram/image
In addition to the co-primary efficacy endpoints, statistically significant improvements were also demonstrated in the following prospectively defined endpoints.
The median time to initiation of cytotoxic chemotherapy was 28.0 months for patients receiving enzalutamide and 10.8 months for patients receiving placebo [(HR = 0.35 (95% CI: [0.30, 0.40)], p < 0.0001].
The proportion of enzalutamide-treated patients with measurable disease at baseline who had an objective soft tissue response was 58.8% (95% CI: 53.8, 63.7) compared with 5.0% (95% CI: 3.0, 7.7) of patients receiving placebo. The absolute difference in objective soft tissue response between enzalutamide and placebo arms was [53.9% (95% CI: 48.5, 59.1), p < 0.0001]. Complete responses were reported in 19.7% of enzalutamide-treated patients compared with 1.0% of placebo-treated patients, and partial responses were reported in 39.1% of enzalutamide-treated patients versus 3.9% of placebo-treated patients.
Enzalutamide significantly decreased the risk of the first skeletal-related event by 28% [HR = 0.72 (95% CI: 0.61, 0.84), p < 0.0001]. A skeletal-related event was defined as radiation therapy or surgery to bone for prostate cancer, pathologic bone fracture, spinal cord compression, or change of antineoplastic therapy to treat bone pain. The analysis included 587 skeletal-related events, of which 389 events (66.3%) were radiation to bone, 79 events (13.5%) were spinal cord compression, 70 events (11.9%) were pathologic bone fracture, 45 events (7.6%) were change in antineoplastic therapy to treat bone pain, and 22 events (3.7%) were surgery to bone.
Patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 78.0% versus 3.5% (difference = 74.5%, p < 0.0001).
The median time to PSA progression per PCWG2 criteria was 11.2 months for patients treated with enzalutamide and 2.8 months for patients who received placebo [HR = 0.17 (95% CI: 0.15, 0.20), p < 0.0001].
Treatment with enzalutamide decreased the risk of FACT-P degradation by 37.5% compared with placebo (p<0.0001). The median time to degradation in FACT-P was 11.3 months in the enzalutamide group and 5.6 months in the placebo group.
CRPC2 (AFFIRM) study (patients with metastatic CRPC who previously received chemotherapy): The efficacy and safety of enzalutamide in patients with metastatic castration-resistant prostate cancer who had received docetaxel and were using a LHRH analogue or had undergone orchiectomy were assessed in a randomised, placebo-controlled, multicentre phase 3 clinical trial. A total of 1199 patients were randomised 2:1 to receive either enzalutamide orally at a dose of 160 mg once daily (N = 800) or placebo once daily (N = 399). Patients were allowed but not required to take prednisone (maximum daily dose allowed was 10 mg prednisone or equivalent). Patients randomized to either arm were to continue treatment until disease progression (defined as confirmed radiographic progression or the occurrence of a skeletal-related event) and initiation of new systemic antineoplastic treatment, unacceptable toxicity, or withdrawal.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 69 years (range 41-92) and the racial distribution was 93% Caucasian, 4% Black, 1% Asian, and 2% Other. The ECOG performance score was 0-1 in 91.5% of patients and 2 in 8.5% of patients; 28% had a mean Brief Pain Inventory score of ≥ 4 (mean of patient's reported worst pain over the previous 24 hours calculated for seven days prior to randomization). Most (91%) patients had metastases in bone and 23% had visceral lung and/or liver involvement. At study entry, 41% of randomized patients had PSA progression only, whereas 59% of patients had radiographic progression. Fifty-one percent (51%) of patients were on bisphosphonates at baseline.
The AFFIRM study excluded patients with medical conditions that may predispose them to seizures (see Adverse Reactions) and medicinal products known to decrease the seizure threshold, as well as clinically significant cardiovascular disease such as uncontrolled hypertension, recent history of myocardial infarction or unstable angina, New York Heart Association class III or IV heart failure (unless ejection fraction was ≥ 45%), clinically significant ventricular arrhythmias or AV block (without permanent pacemaker).
The protocol pre-specified interim analysis after 520 deaths showed a statistically significant superiority in overall survival in patients treated with enzalutamide compared to placebo (see Table 4 and Figures 12 and 13).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In addition to the observed improvement in overall survival, key secondary endpoints (PSA progression, radiographic progression-free survival, and time to first skeletal-related event) favoured enzalutamide and were statistically significant after adjusting for multiple testing.
Radiographic progression-free survival as assessed by the investigator using RECIST v1.1 for soft tissue and appearance of 2 or more bone lesions in bone scan was 8.3 months for patients treated with enzalutamide and 2.9 months for patients who received placebo [HR = 0.40, (95% CI: 0.35, 0.47]), p < 0.0001]. The analysis involved 216 deaths without documented progression and 645 documented progression events, of which 303 (47%) were due to soft tissue progression, 268 (42%) were due to bone lesion progression and 74 (11%) were due to both soft tissue and bone lesions.
Confirmed PSA decline of 50% or 90% were 54.0% and 24.8%, respectively, for patients treated with enzalutamide and 1.5% and 0.9%, respectively, for patients who received placebo (p < 0.0001). The median time to PSA progression was 8.3 months for patients treated with enzalutamide and 3.0 months for patients who received placebo [HR = 0.25, (95% CI: 0.20, 0.30), p < 0.0001].
The median time to first skeletal-related event was 16.7 months for patients treated with enzalutamide and 13.3 months for patients who received placebo [HR = 0.69 (95% CI: 0.57, 0.84), p < 0.0001]. A skeletal-related event was defined as radiation therapy or surgery to bone, pathologic bone fracture, spinal cord compression, or change of antineoplastic therapy to treat bone pain. The analysis involved 448 skeletal-related events, of which 277 events (62%) were radiation to bone, 95 events (21%) were spinal cord compression, 47 events (10%) were pathologic bone fracture, 36 events (8%) were change in antineoplastic therapy to treat bone pain and 7 events (2%) were surgery to bone.
Elderly: Of the 4168 patients in the controlled clinical trials who received enzalutamide, 3265 patients (78%) were 65 years and over and 1469 patients (35%) were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients.
Pharmacokinetics: Enzalutamide is poorly water soluble. In this product, the solubility of enzalutamide is increased by caprylocaproyl macrogolglycerides as emulsifier/surfactant. In preclinical studies, the absorption of enzalutamide was increased when dissolved in caprylocaproyl macrogolglycerides.
The pharmacokinetics of enzalutamide have been evaluated in prostate cancer patients and in healthy male subjects. The mean terminal half-life (t
½) for enzalutamide in patients after a single oral dose is 5.8 days (range 2.8 to 10.2 days), and steady state is achieved in approximately one month. With daily oral administration, enzalutamide accumulates approximately 8.3-fold relative to a single dose. Daily fluctuations in plasma concentrations are low (peak-to-trough ratio of 1.25). Clearance of enzalutamide is primarily via hepatic metabolism, producing an active metabolite that is equally as active as enzalutamide and circulates at approximately the same plasma concentration as enzalutamide.
Absorption: Maximum plasma concentrations (C
max) of enzalutamide in patients are observed 1 to 2 hours after administration. Based on a mass balance study in humans, oral absorption of enzalutamide is estimated to be at least 84.2%. Enzalutamide is not a substrate of the efflux transporters P-gp or BCRP. At steady state, the mean C
max values for enzalutamide and its active metabolite are 16.6 μg/mL (23% coefficient of variation [CV]) and 12.7 μg/mL (30% CV), respectively.
Food has no clinically significant effect on the extent of absorption. In clinical trials, Xtandi was administered without regard to food.
Distribution: The mean apparent volume of distribution (V/F) of enzalutamide in patients after a single oral dose is 110 L (29% CV). The volume of distribution of enzalutamide is greater than the volume of total body water, indicative of extensive extravascular distribution. Studies in rodents indicate that enzalutamide and its active metabolite can cross the blood brain barrier.
Enzalutamide is 97% to 98% bound to plasma proteins, primarily albumin. The active metabolite is 95% bound to plasma proteins. There was no protein binding displacement between enzalutamide and other highly bound drugs (warfarin, ibuprofen and salicylic acid)
in vitro.
Biotransformation: Enzalutamide is extensively metabolized. There are two major metabolites in human plasma: N-desmethyl enzalutamide (active) and a carboxylic acid derivative (inactive). Following single oral administration of
14C-enzalutamide 160 mg, plasma samples were analyzed for enzalutamide and its metabolites up to 77 days post dose. Enzalutamide, N-desmethyl enzalutamide, and a major inactive carboxylic acid metabolite accounted for 88% of the
14C-radioactivity in plasma, representing 30%, 49%, and 10%, respectively, of the total
14C-AUC
0-inf.
Enzalutamide is metabolized by CYP2C8 and to a lesser extent by CYP3A4/5 (see Interactions), both of which play a role in the formation of the active metabolite.
In vitro, N-desmethyl enzalutamide is metabolized to the carboxylic acid metabolite by carboxylesterase 1, which also plays a minor role in the metabolism of enzalutamide to the carboxylic acid metabolite. Carboxylesterase 2 does not appear to play a role in the metabolism of either enzalutamide or N-desmethyl enzalutamide. N-desmethyl enzalutamide was not metabolized by CYPs
in vitro.
Under conditions of clinical use, enzalutamide is a strong inducer of CYP3A4, a moderate inducer of CYP2C9 and CYP2C19, and has no clinically relevant effect on CYP2C8 (see Interactions).
Elimination: The mean apparent clearance (CL/F) of enzalutamide in patients ranges from 0.520 and 0.564 L/h.
Following oral administration of
14C-enzalutamide, 84.6% of the radioactivity is recovered by 77 days post dose: 71.0% is recovered in urine (primarily as the inactive metabolite, with trace amounts of enzalutamide and the active metabolite), and 13.6% is recovered in faeces (0.39% of dose as unchanged enzalutamide).
In vitro data indicate that enzalutamide is not a substrate for OATP1B1, OATP1B3, or OCT1; and N-desmethyl enzalutamide is not a substrate for P-gp or BCRP.
In vitro data indicate that enzalutamide and its major metabolites do not inhibit the following transporters at clinically relevant concentrations: OATP1B1, OATP1B3, OCT2, or OAT1.
Linearity: No major deviations from dose proportionality are observed over the dose range 40 to 160 mg. The steady-state C
min values of enzalutamide and the active metabolite in individual patients remained constant during more than one year of chronic therapy, demonstrating time-linear pharmacokinetics once steady-state is achieved.
Renal impairment: No formal renal impairment study for enzalutamide has been completed. Patients with serum creatinine > 177 μmol/L (2 mg/dL) were excluded from clinical studies. Based on a population pharmacokinetic analysis, no dose adjustment is necessary for patients with calculated creatinine clearance (CrCL) values ≥ 30 mL/min (estimated by the Cockcroft and Gault formula). Enzalutamide has not been evaluated in patients with severe renal impairment (CrCL < 30 mL/min) or end-stage renal disease, and caution is advised when treating these patients. It is unlikely that enzalutamide will be significantly removed by intermittent haemodialysis or continuous ambulatory peritoneal dialysis.
Hepatic impairment: The pharmacokinetics of enzalutamide were examined in subjects with baseline mild (N = 6), moderate (N = 8), or severe (N = 8) hepatic impairment (Child-Pugh Class A, B or C, respectively) and in 22 matched control subjects with normal hepatic function. Following a single oral 160 mg dose of enzalutamide, the AUC and C
max for enzalutamide in subjects with mild impairment increased by 5% and 24%, respectively, the AUC and C
max of enzalutamide in subjects with moderate impairment increased by 29% and decreased by 11%, respectively, and the AUC and C
max of enzalutamide in subjects with severe impairment increased by 5% and decreased by 41%, respectively, compared to healthy control subjects. For the sum of unbound enzalutamide plus the unbound active metabolite, the AUC and C
max in subjects with mild impairment increased by 14% and 19%, respectively, the AUC and C
max in subjects with moderate impairment increased by 14% and decreased by 17%, respectively, and the AUC and C
max in subjects with severe hepatic impairment increased by 34% and decreased by 27%, respectively, compared to healthy control subjects.
Race: Most patients in the randomized clinical studies (> 77%) were Caucasian. Based on pharmacokinetic data from studies in Japanese and Chinese patients with prostate cancer, there were no clinically relevant differences in exposure among the populations. There are insufficient data to evaluate potential differences in the pharmacokinetics of enzalutamide in other races.
Elderly: No clinically relevant effect of age on enzalutamide pharmacokinetics was seen in the elderly population pharmacokinetic analysis.
Toxicology: Preclinical safety data: In a 6-month study in transgenic rasH2 mice, enzalutamide did not show carcinogenic potential (absence of neoplastic findings) at doses up to 20 mg/kg per day (AUC
24h 317 μg.h/mL), which resulted in plasma exposure levels similar to the clinical exposure (AUC
24h 322 μg.h/mL) in mCRPC patients receiving 160 mg, daily. Daily oral dosing of rats with enzalutamide at 10 to 100 mg/kg for 2 years increased the incidence of neoplastic findings (compared to control) that were considered related to the primary pharmacology of enzalutamide. These included benign thymoma, fibroadenoma in the mammary glands, and benign Leydig cell tumors in the testes in males; benign granulosa cell tumor in the ovaries in females; and adenoma in the pars distalis of the pituitary in both sexes. In addition, urothelial papilloma and carcinoma of urinary bladder in male rats were observed at the 100 mg/kg/day dose and were considered secondary to the irritation caused by the increased urinary crystal/calculi, which is known to occur in rodent species. Leydig cell tumors in rats are generally not considered relevant to humans based on experience with other anti-androgens. The human relevance of thymoma, pituitary adenoma and fibroadenoma in rats is unclear, but a potential relevance cannot be ruled out. The exposure levels (based on AUC) achieved in this study, for enzalutamide and its metabolites, M1 and M2, in rats were less than or similar to those in prostate cancer patients at the recommended dose of enzalutamide.
Enzalutamide did not induce mutations in the bacterial reverse mutation (Ames) assay, was non-mutagenic, non-clastogenic in mammalian cells, and non-genotoxic
in vivo in mice. Enzalutamide did not induce phototoxicity in cultured mammalian cells.
Enzalutamide could cause fetal harm when administered to a pregnant woman based on its mechanism of action and embryo-fetal toxicity observed in mice. Based on nonclinical findings in repeat-dose toxicology studies, which were consistent with the pharmacological activity of enzalutamide, male fertility may be impaired by treatment with enzalutamide. In studies in mice (4 weeks), rats (4 and 26 weeks), and dogs (4, 13, and 39 weeks), changes in the reproductive organs associated with enzalutamide were decreases in organ weight with atrophy of the prostate and epididymis.
In a pharmacokinetic study in pregnant rats with a single oral 30 mg/kg enzalutamide administration on gestation day 14, enzalutamide and/or its metabolites were present in the fetus at a C
max that was approximately 0.3 times the concentration found in maternal plasma and occurred 4 hours after administration.
Following a single oral administration in lactating rats on postnatal day 14, enzalutamide and/or its metabolites were present in milk at a C
max that was 4 times higher than concentrations in the plasma and occurred 4 hours after administration.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image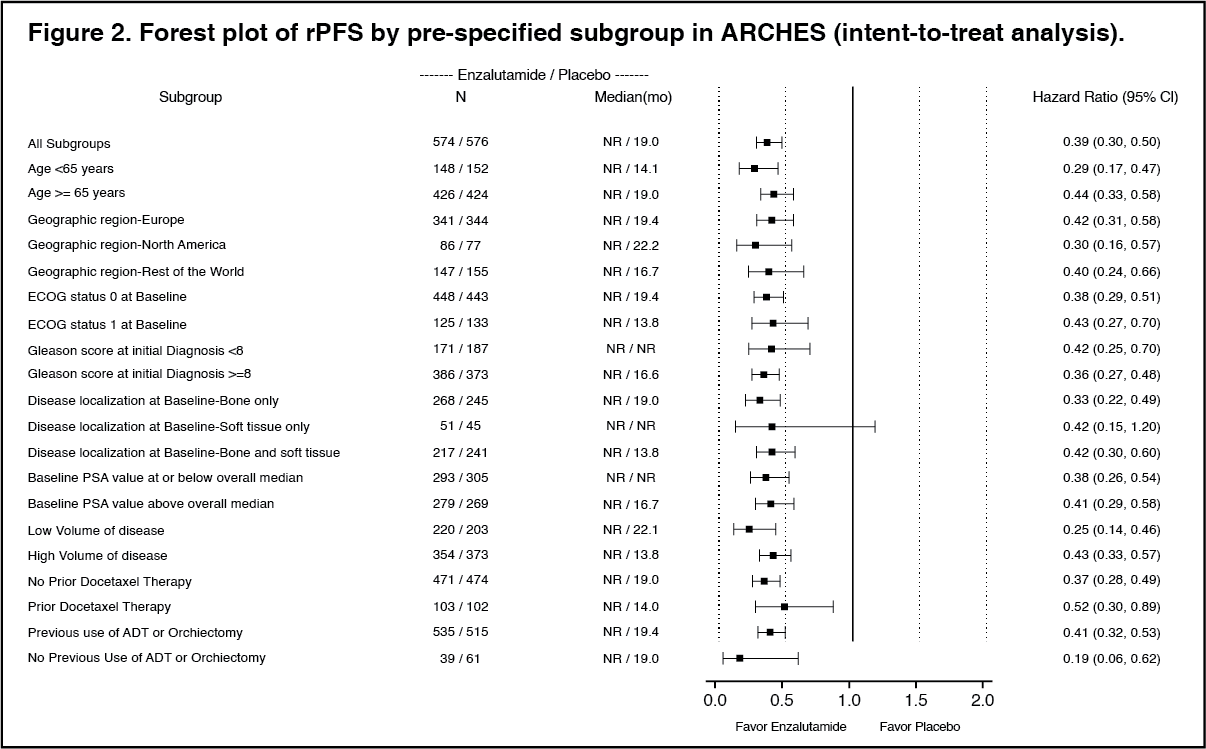 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image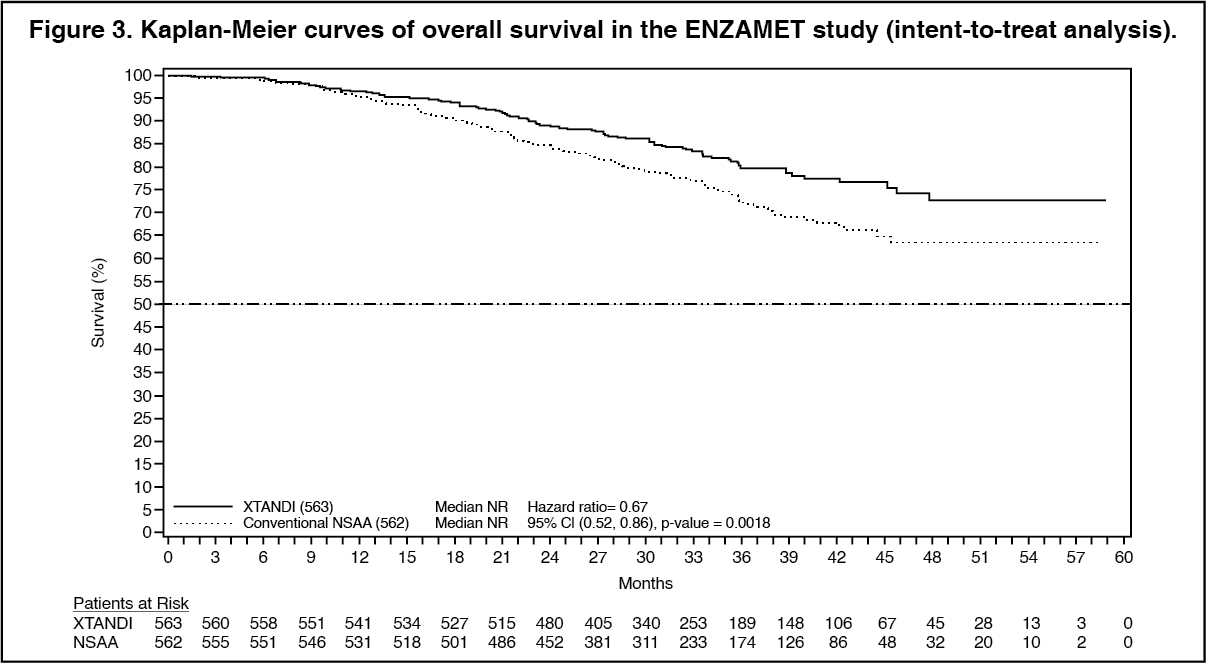 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image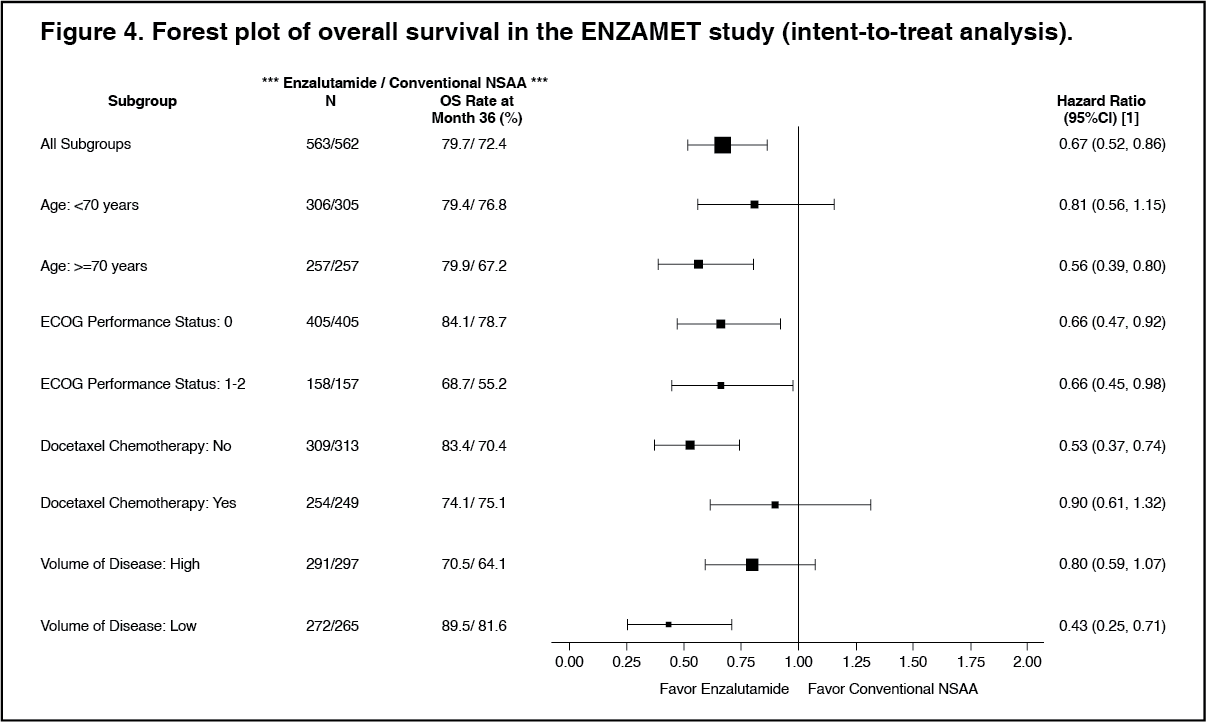 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image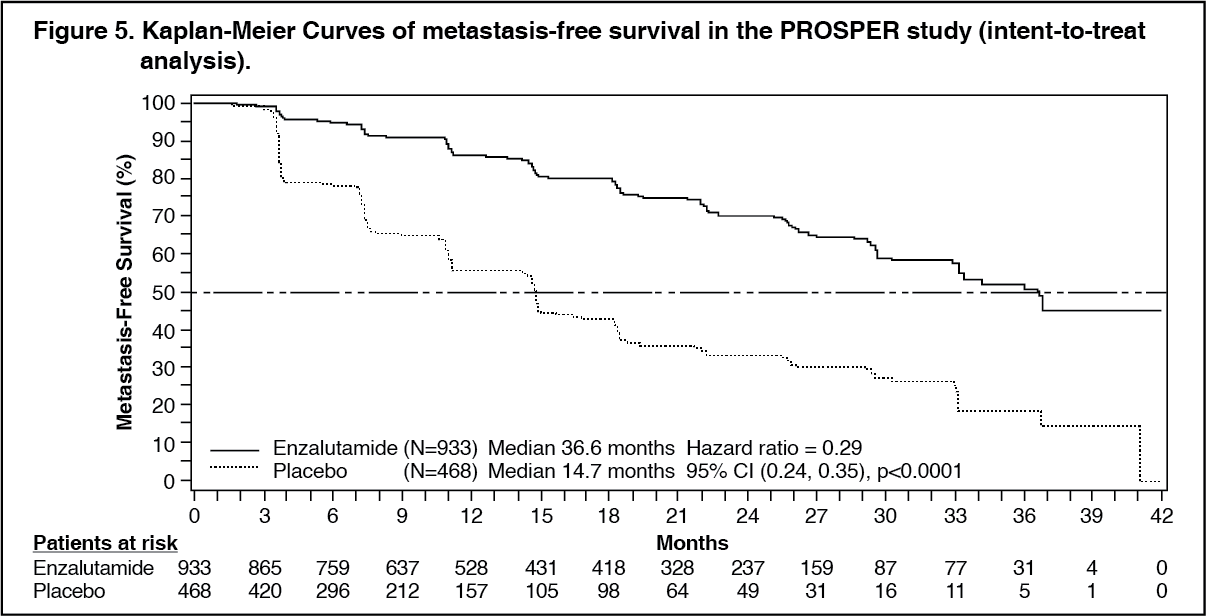 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image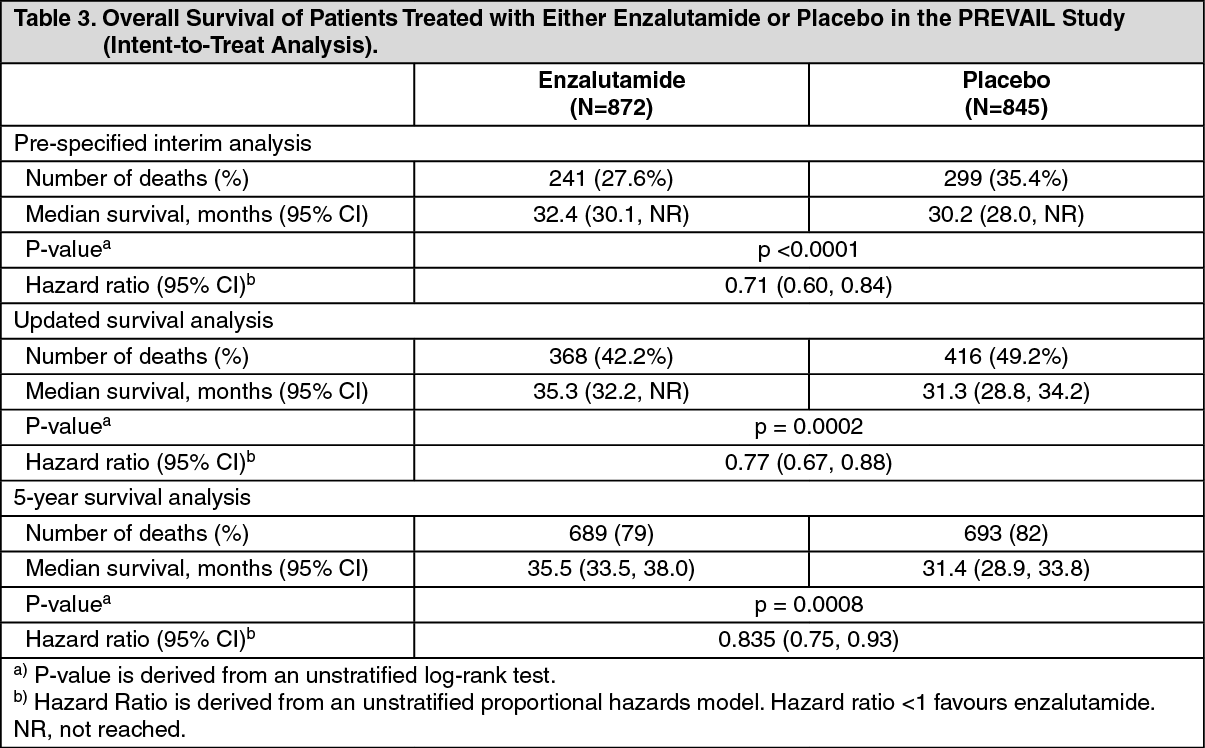 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image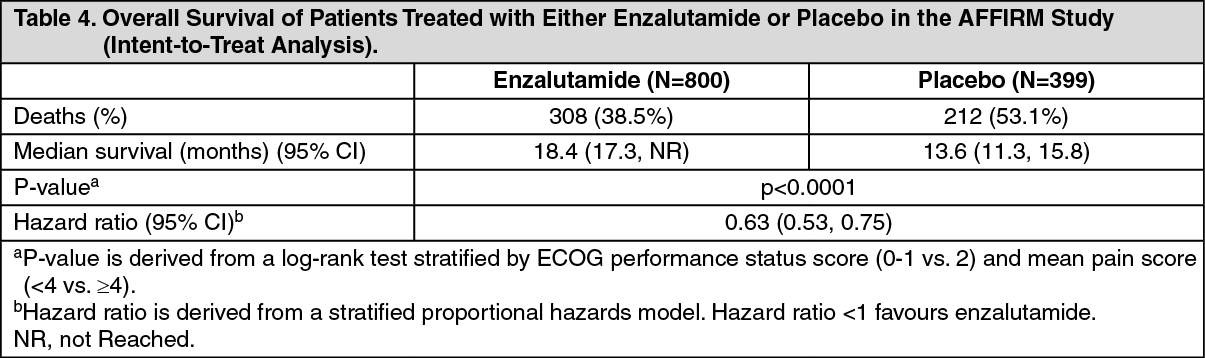 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
