Pharmacology: Mechanism of Action: Emtricitabine:
Emtricitabine, a synthetic nucleoside analog of cytidine, is phosphorylated by cellular enzymes to form emtricitabine 5'-triphosphate. Emtricitabine 5'-triphosphate inhibits the activity of the HIV-1 reverse transcriptase (RT) by competing with the natural substrate deoxycytidine 5'-triphosphate and by being incorporated into nascent viral DNA which results in chain termination. Emtricitabine 5'-triphosphate is a weak inhibitor of mammalian DNA polymerase α, β, ε, and mitochondrial DNA polymerase γ.
Tenofovir disoproxil fumarate: Tenofovir disoproxil fumarate is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. Tenofovir disoproxil fumarate requires initial diester hydrolysis for conversion to tenofovir, and subsequent phosphorylations by cellular enzymes, to form tenofovir diphosphate. Tenofovir diphosphate inhibits the activity of HIV-1 RT by competing with the natural substrate deoxyadenosine 5'-triphosphate and, after incorporation into DNA, by DNA-chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Antiviral Activity: Emtricitabine and tenofovir disoproxil fumarate: In combination studies evaluating the in cell culture antiviral activity of emtricitabine and tenofovir together, synergistic antiviral effects were observed.
Emtricitabine: The antiviral activity of emtricitabine against laboratory and clinical isolates of HIV was assessed in lymphoblastoid cell lines, the MAGI-CCR5 cell line, and peripheral blood mononuclear cells. The 50% effective concentration (EC
50) values for emtricitabine were in the range of 0.0013–0.64 μM (0.0003–0.158 μg/mL). In drug combination studies of emtricitabine with nucleoside reverse transcriptase inhibitors (abacavir, lamivudine, stavudine, zalcitabine, zidovudine), non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, nelfinavir, ritonavir, saquinavir), additive to synergistic effects were observed. Emtricitabine displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC
50 values ranged from 0.007–0.075 μM) and showed strain specific activity against HIV-2 (EC
50 values ranged from 0.007–1.5 μM).
Tenofovir disoproxil fumarate: The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC
50 values for tenofovir were in the range of 0.04–8.5 μM. In drug combination studies of tenofovir with nucleoside reverse transcriptase inhibitors (abacavir, didanosine, lamivudine, stavudine, zalcitabine, zidovudine), non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir), additive to synergistic effects were observed. Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G and O (EC
50 values ranged from 0.5–2.2 μM) and showed strain specific activity against HIV-2 (EC
50 values ranged from 1.6 μM to 4.9 μM).
Resistance: Emtricitabine and tenofovir disoproxil fumarate: HIV-1 isolates with reduced susceptibility to the combination of emtricitabine and tenofovir have been selected in cell culture. Genotypic analysis of these isolates identified the M184V/I and/or K65R amino acid substitutions in the viral RT.
In a clinical study of treatment-naïve patients (Study 934, see Description of Clinical Studies under INDICATIONS/USES) resistance analysis was performed on HIV isolates from all virologic failure patients with >400 copies/mL of HIV-1 RNA at Week 48 or early discontinuations. Development of efavirenz resistance-associated mutations occurred most frequently and was similar between the treatment arms. The M184V amino acid substitution, associated with resistance to EMTRIVA and lamivudine, was observed in 2/12 (17%) analyzed patient isolates in the EMTRIVA + VIREAD group and in 7/22 (32%) analyzed patient isolates in the zidovudine/lamivudine group. Through 48 weeks of Study 934, no patients have developed a detectable K65R mutation in their HIV as analyzed through standard genotypic analysis. Insufficient data are available to assess the development of the K65R mutation upon prolonged exposure to this regimen.
Emtricitabine: Emtricitabine-resistant isolates of HIV have been selected in cell culture and in vivo. Genotypic analysis of these isolates showed that the reduced susceptibility to emtricitabine was associated with a mutation in the HIV RT gene at codon 184 which resulted in an amino acid substitution of methionine by valine or isoleucine (M184V/I).
Tenofovir disoproxil fumarate: HIV-1 isolates with reduced susceptibility to tenofovir have been selected in cell culture. These viruses expressed a K65R mutation in RT and showed a 2-4 fold reduction in susceptibility to tenofovir.
In treatment-naïve patients, isolates from 8 patients developed the K65R mutation in the VIREAD arm through 144 weeks; 7 occurred in the first 48 weeks of treatment and 1 at Week 96. In treatment-experienced patients, 14/304 (5%) isolates from patients failing VIREAD through Week 96 showed >1.4 fold (median 2.7) reduced susceptibility to tenofovir. Genotypic analysis of the resistant isolates showed a mutation in the HIV-1RT gene resulting in the K65R amino acid substitution.
Cross-resistance: Emtricitabine and tenofovir disoproxil fumarate: Cross-resistance among certain nucleoside reverse transcriptase inhibitors (NRTIs) has been recognized. The M184V/I and/or K65R substitutions selected in cell culture by the combination of emtricitabine and tenofovir are also observed in some HIV-1 isolates from subjects failing treatment with tenofovir in combination with either lamivudine or emtricitabine, and either abacavir or didanosine. Therefore, cross-resistance among these drugs may occur in patients whose virus harbors either or both of these amino acid substitutions.
Emtricitabine: Emtricitabine-resistant isolates (M184V/I) were cross-resistant to lamivudine and zalcitabine but retained susceptibility in cell culture to didanosine, stavudine, tenofovir, zidovudine, and NNRTIs (delavirdine, efavirenz, and nevirapine). HIV-1 isolates containing the K65R substitution, selected in vivo by abacavir, didanosine, tenofovir, and zalcitabine, demonstrated reduced susceptibility to inhibition by emtricitabine. Viruses harboring mutations conferring reduced susceptibility to stavudine and zidovudine (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E), or didanosine (L74V) remained sensitive to emtricitabine. HIV-1 containing the K103N substitution associated with resistance to NNRTIs was susceptible to emtricitabine.
Tenofovir disoproxil fumarate: HIV-1 isolates from patients (N=20) whose HIV-1expressed a mean of 3 zidovudine-associated RT amino acid substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir. Multinucleoside resistant HIV-1 with a T69S double insertion mutation in the RT showed reduced susceptibility to tenofovir.
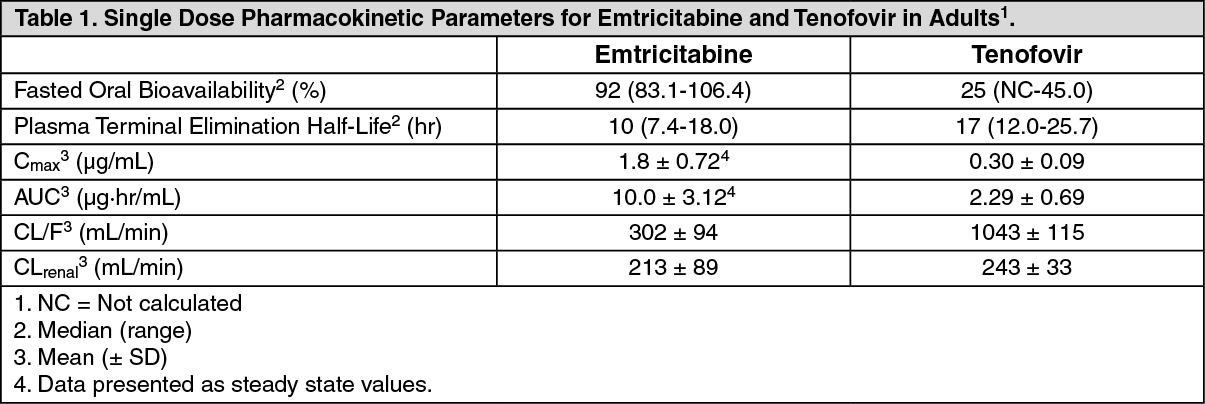
Pharmacokinetics: Pharmacokinetics in Adults: TRUVADA: One TRUVADA Tablet was bioequivalent to one EMTRIVA Capsule (200 mg) plus one VIREAD Tablet (300 mg) following single-dose administration to fasting healthy subjects (N=39).
Emtricitabine: The pharmacokinetic properties of emtricitabine are summarized in Table 1. Following oral administration of EMTRIVA, emtricitabine is rapidly absorbed with peak plasma concentrations occurring at 1-2 hours post-dose. In vitro binding of emtricitabine to human plasma proteins is <4% and is independent of concentration over the range of 0.02-200 μg/mL. Following administration of radiolabelled emtricitabine, approximately 86% is recovered in the urine and 13% is recovered as metabolites. The metabolites of emtricitabine include 3'-sulfoxide diastereomers and their glucuronic acid conjugate. Emtricitabine is eliminated by a combination of glomerular filtration and active tubular secretion. Following a single oral dose of EMTRIVA, the plasma emtricitabine half-life is approximately 10 hours.
Tenofovir disoproxil fumarate: The pharmacokinetic properties of tenofovir disoproxil fumarate are summarized in Table 1. Following oral administration of VIREAD, maximum tenofovir serum concentrations are achieved in 1.0 ± 0.4 hour. In vitro binding of tenofovir to human plasma proteins is <0.7% and is independent of concentration over the range of 0.01-25 μg/mL. Approximately 70-80% of the intravenous dose of tenofovir is recovered as unchanged drug in the urine. Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion. Following a single oral dose of VIREAD, the terminal elimination half-life of tenofovir is approximately 17 hours. (See Table 1.)
 Click on icon to see table/diagram/image
Effects of Food on Oral Absorption:
Click on icon to see table/diagram/image
Effects of Food on Oral Absorption: TRUVADA may be administered with or without food. Administration of TRUVADA following a high fat meal (784 kcal; 49 grams of fat) or a light meal (373 kcal; 8 grams of fat) delayed the time of tenofovir C
max by approximately 0.75 hr. The mean increases in tenofovir AUC and C
max were approximately 35% and 15% respectively, when administered with a high fat or light meal, compared to administration in the fasted state. In previous safety and efficacy studies, VIREAD (tenofovir) was taken under fed conditions. Emtricitabine systemic exposures (AUC and C
max) were unaffected when TRUVADA was administered with either a high fat or a light meal.
Special Populations: Race: Emtricitabine: No pharmacokinetic differences due to race have been identified following the administration of EMTRIVA.
Tenofovir disoproxil fumarate: There were insufficient numbers from racial and ethnic groups other than Caucasian to adequately determine potential pharmacokinetic differences among these populations following the administration of VIREAD.
Gender:
Emtricitabine and tenofovir disoproxil fumarate: Emtricitabine and tenofovir pharmacokinetics are similar in male and female patients.
Pediatric and Geriatric Patients: Pharmacokinetic studies of tenofovir have not been performed in pediatric patients (<18 years). Pharmacokinetics of emtricitabine and tenofovir have not been fully evaluated in the elderly (>65 years) (see Use in Children and Use in the Elderly under PRECAUTIONS).
Patients with Impaired Renal Function: The pharmacokinetics of emtricitabine and tenofovir are altered in patients with renal impairment (see Renal Impairment under WARNINGS). In patients with creatinine clearance <50 mL/min, C
max, and AUC
0-∞ of emtricitabine and tenofovir were increased. It is recommended that the dosing interval for TRUVADA be modified in patients with creatinine clearance 30-49 mL/min. TRUVADA should not be used in patients with creatinine clearance <30 mL/min and in patients with end-stage renal disease requiring dialysis (see DOSAGE & ADMINISTRATION).
Patients with Hepatic Impairment: The pharmacokinetics of tenofovir following a 300 mg dose of VIREAD have been studied in non-HIV infected patients with moderate to severe hepatic impairment. There were no substantial alterations in tenofovir pharmacokinetics in patients with hepatic impairment compared with unimpaired patients. The pharmacokinetics of TRUVADA or emtricitabine have not been studied in patients with hepatic impairment; however, emtricitabine is not significantly metabolized by liver enzymes, so the impact of liver impairment should be limited.
Pregnancy: (see Use in Pregnancy & Lactation).
Nursing Mothers: (see Use in Pregnancy & Lactation).
Drug Interactions (see Drug Interactions under PRECAUTIONS)
: TRUVADA: No drug interaction studies have been conducted using TRUVADA Tablets.
Emtricitabine and tenofovir disoproxil fumarate:
The steady state pharmacokinetics of emtricitabine and tenofovir were unaffected, when emtricitabine and tenofovir disoproxil fumarate were administered together versus each agent dosed alone.
In vitro and clinical pharmacokinetic drug-drug interaction studies have shown that the potential for CYP450 mediated interactions involving emtricitabine and tenofovir with other medicinal products is low.
Emtricitabine and tenofovir are primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion. No drug-drug interactions due to competition for renal excretion have been observed; however, co-administration of TRUVADA with drugs that are eliminated by active tubular secretion may increase concentrations of emtricitabine, tenofovir, and/or the co-administered drug.
Drugs that decrease renal function may increase concentrations of emtricitabine and/or tenofovir.
No clinically significant drug interactions have been observed between emtricitabine and famciclovir, indinavir, stavudine, tenofovir disoproxil fumarate, and zidovudine (see Tables 2 and 3). Similarly, no clinically significant drug interactions have been observed between tenofovir disoproxil fumarate and abacavir, adefovir dipivoxil, efavirenz, emtricitabine, indinavir, lamivudine, lopinavir/ritonavir, methadone, nelfinavir, oral contraceptives, ribavirin, and saquinavir/ritonavir in studies conducted in healthy volunteers (Tables 4 and 5). (See Tables 2, 3, 4, and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Following multiple dosing to HIV-negative subjects receiving either chronic methadone maintenance therapy or oral contraceptives, or single doses of ribavirin, steady-state tenofovir pharmacokinetics were similar to those observed in previous studies, indicating lack of clinically significant drug interactions between these agents and VIREAD.
Coadministration of tenofovir disoproxil fumarate with didanosine results in changes in the pharmacokinetics of didanosine that may be of clinical significance. Table 6 summarizes the effects of tenofovir disoproxil fumarate on the pharmacokinetics of didanosine. Concomitant dosing of tenofovir disoproxil fumarate with didanosine buffered tablets or enteric-coated capsules significantly increases the C
max and AUC of didanosine. When didanosine 250-mg enteric-coated capsules were administered with tenofovir disoproxil fumarate, systemic exposures of didanosine were similar to those seen with the 400-mg enteric-coated capsules alone under fasted conditions. The mechanism of this interaction is unknown. (see Table 6.)
Click on icon to see table/diagram/image
Microbiology: For additional information on Mechanism of Action, Antiviral Activity, Resistance and Cross Resistance, please consult the EMTRIVA and VIREAD prescribing information.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image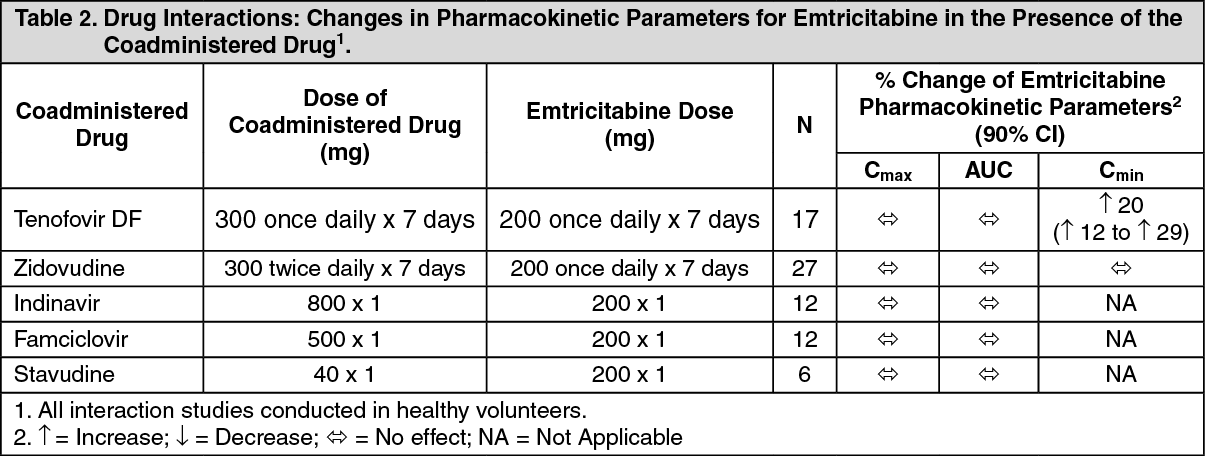 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image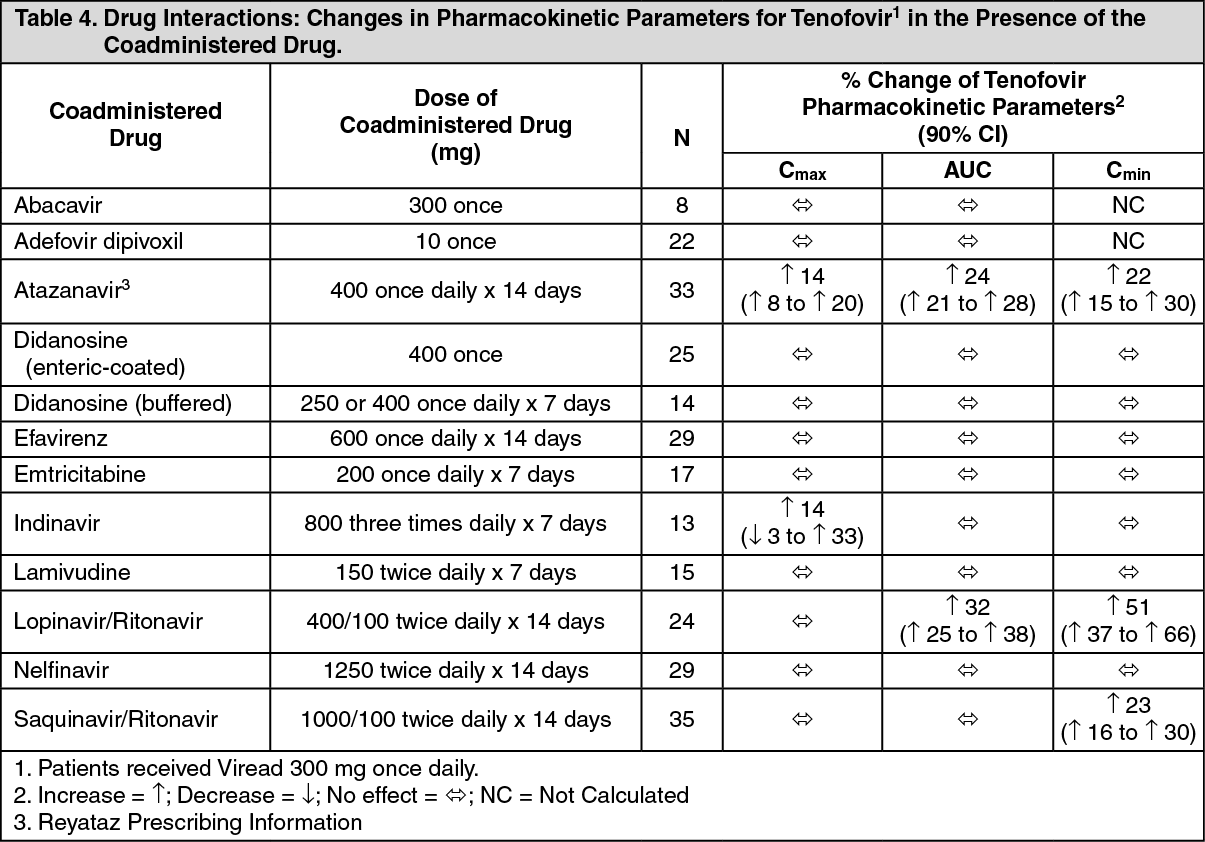 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image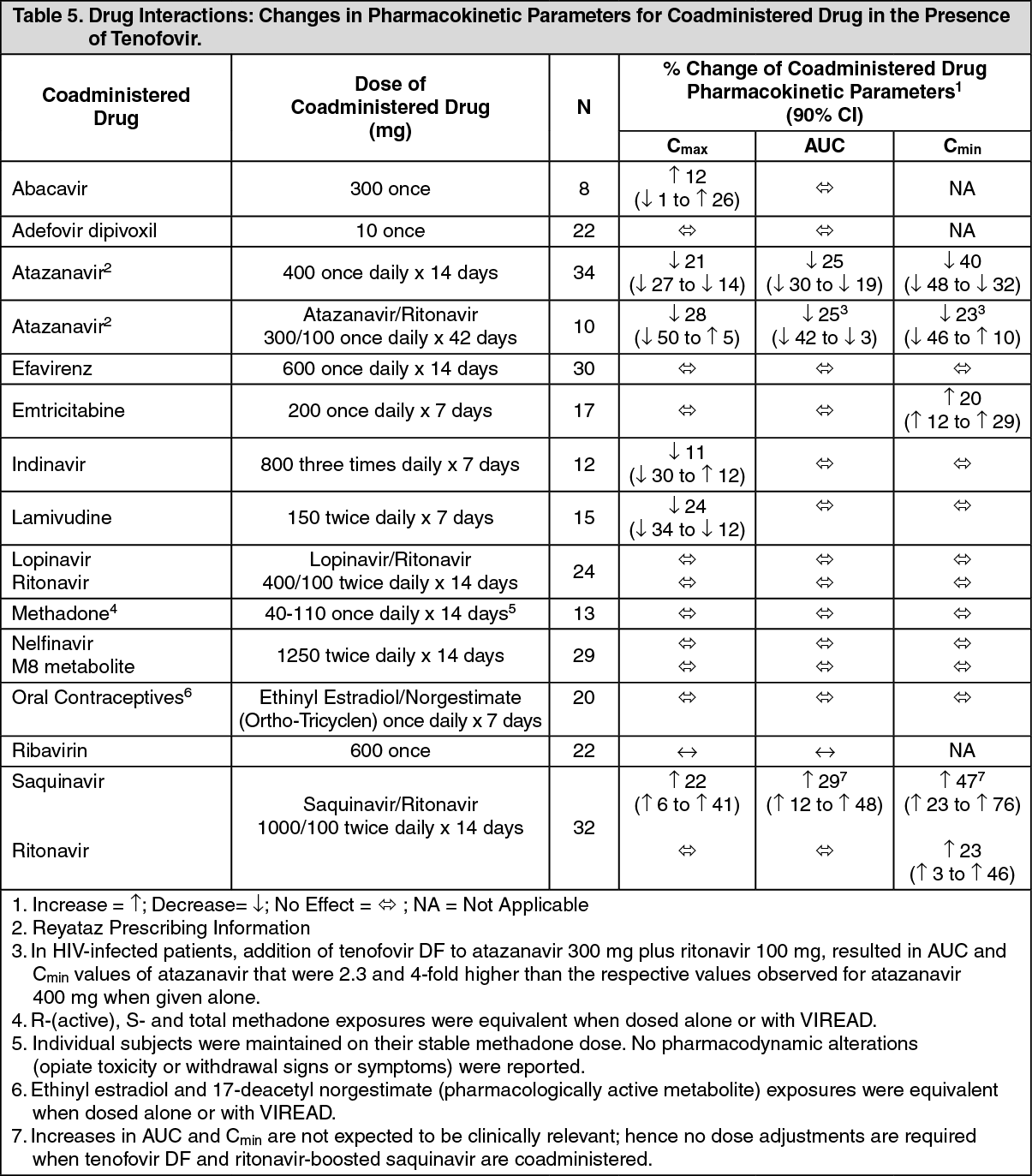 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image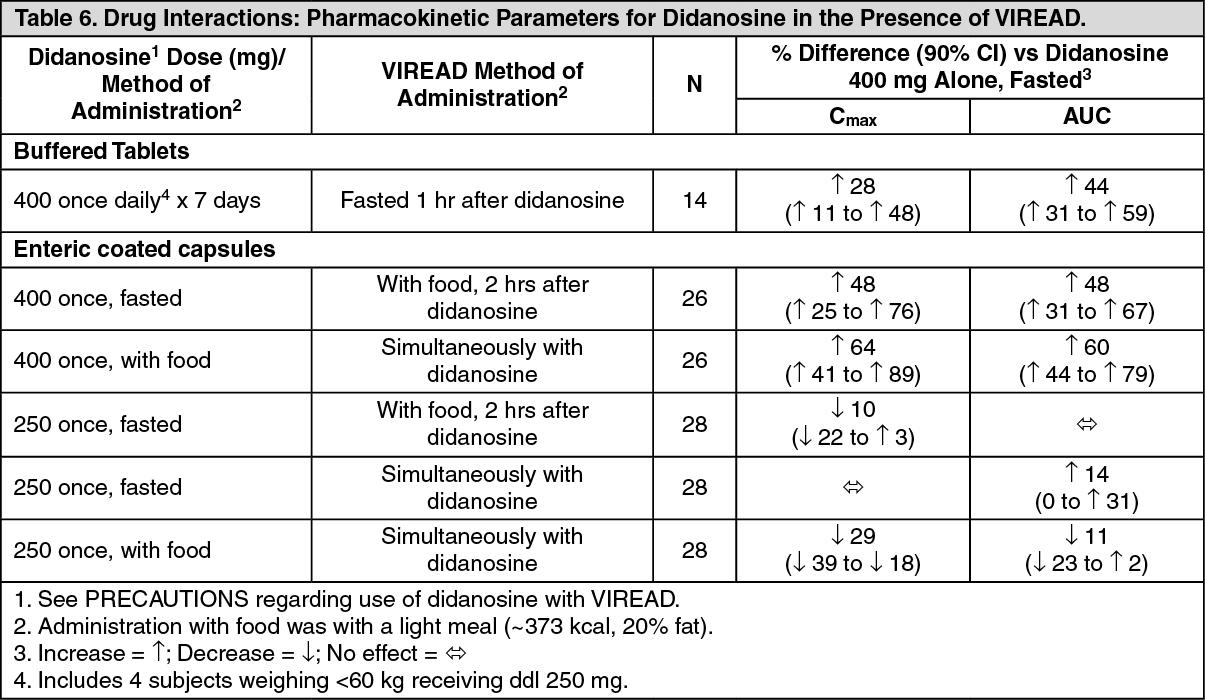 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image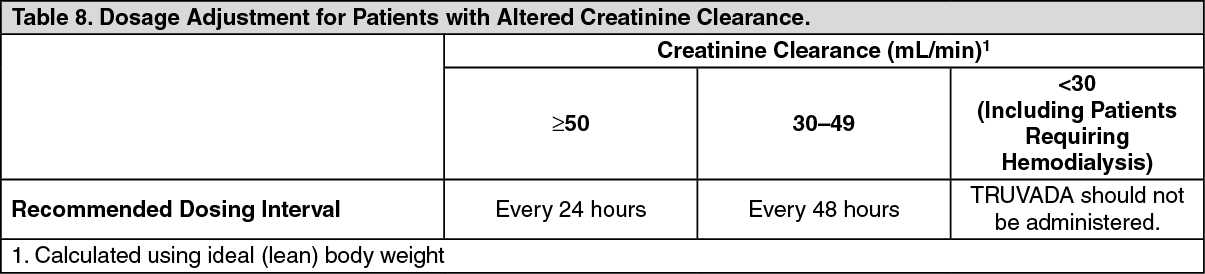 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image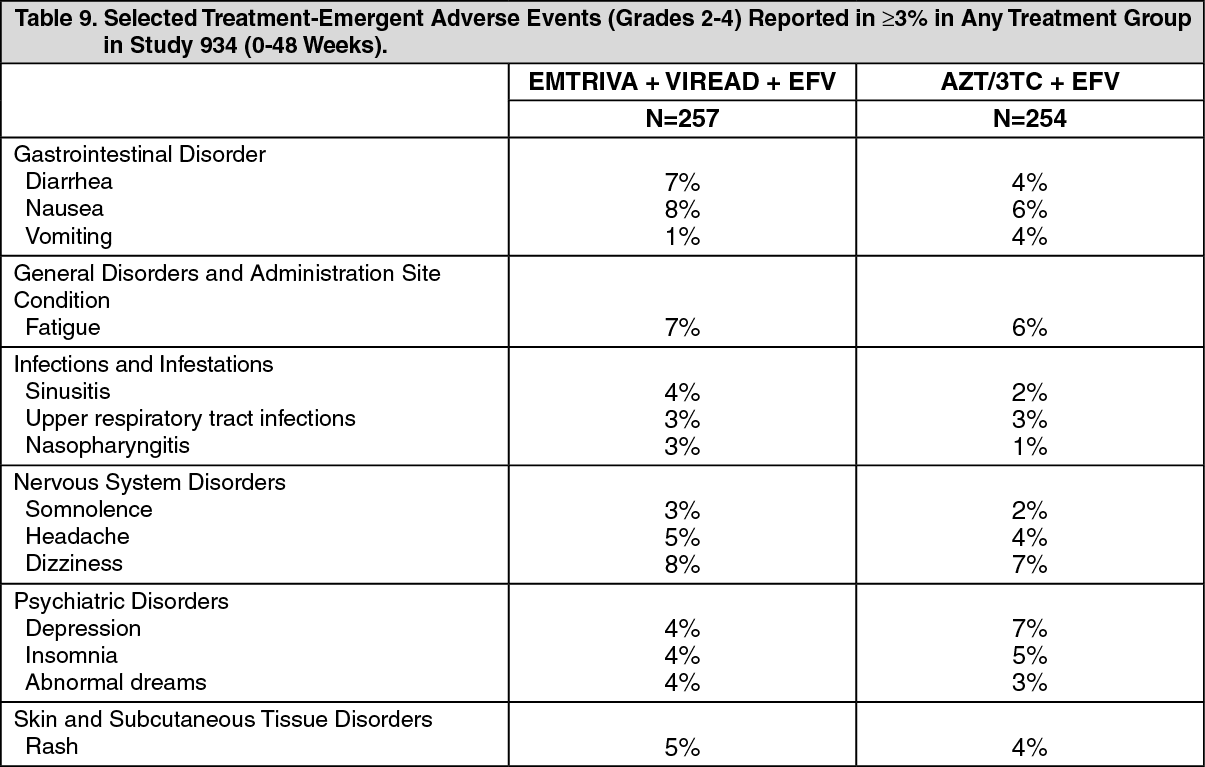 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
