Pharmacology: Pharmacodynamics: Mechanism of action: Capmatinib is a kinase inhibitor that targets MET, including the mutant variant produced by exon 14 skipping. MET exon 14 skipping results in a protein with a missing regulatory domain that reduces its negative regulation leading to increased downstream MET signaling. Capmatinib inhibited cancer cell growth driven by a mutant MET variant lacking exon 14 at clinically achievable concentrations and demonstrated anti-tumor activity in murine tumor xenograft models derived from human lung tumors with either a mutation leading to MET exon 14 skipping or MET amplification. Capmatinib inhibited the phosphorylation of MET triggered by binding of hepatocyte growth factor or by MET amplification, as well as MET-mediated phosphorylation of downstream signaling proteins and proliferation and survival of MET-dependent cancer cells.
Exposure-Response: Capmatinib exposure-response relationships and the time course of pharmacodynamics response are unknown.
Cardiac Electrophysiology: No large mean increase in QTc (i.e. > 20 ms) was detected following treatment with Tabrecta at the recommended dosage of 400 mg orally twice daily.
Clinical efficacy and safety: Metastatic NSCLC with a Mutation that Leads to MET Exon 14 Skipping: The efficacy of Tabrecta was evaluated in GEOMETRY mono-1, a multicenter, non-randomized, open-label, multi-cohort study (NCT02414139). Eligible patients were required to have NSCLC with a mutation that leads to MET exon 14 skipping, epidermal growth factor receptor (EGFR) wild-type and anaplastic lymphoma kinase (ALK) negative status, and at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Patients with symptomatic CNS metastases, clinically significant uncontrolled cardiac disease, or who received treatment with any MET or hepatocyte growth factor (HGF) inhibitor were not eligible for the study.
Out of the 97 patients enrolled in GEOMETRY mono-1 following the central confirmation of MET exon 14 skipping by a RNA-based clinical trial assay, 78 patient samples were retested with the FDA-approved FoundationOne CDx (22 treatment-naïve and 56 previously treated patients) to detect mutations that lead to MET exon 14 skipping. Out of 78 samples retested with FoundationOne CDx, 73 samples were evaluable (20 treatment-naïve and 53 previously treated patients), 72 (20 treatment-naïve and 52 previously treated patients) of which were confirmed to have a mutation that leads to MET exon 14 skipping, demonstrating an estimated positive percentage agreement of 99% (72/73) between the clinical trial assay and the FDA-approved assay.
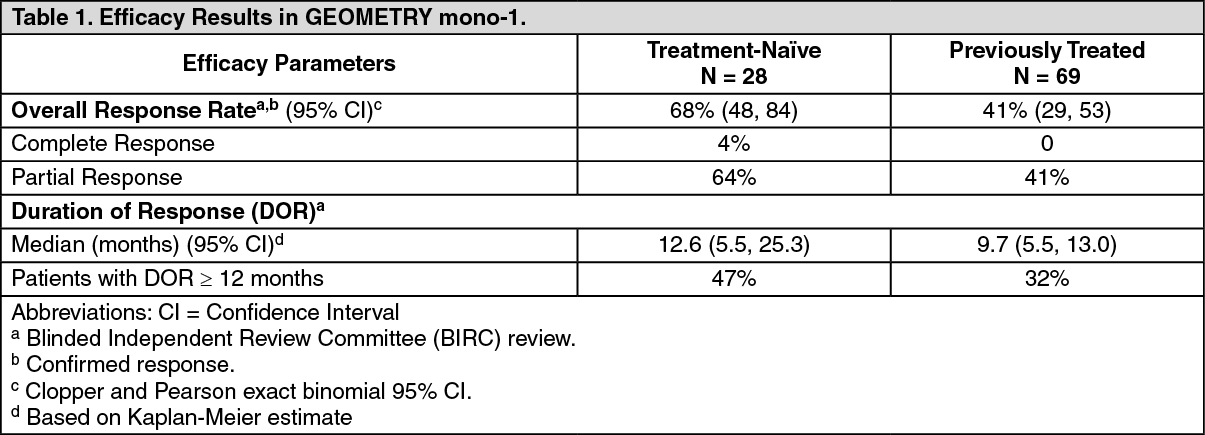
Patients received Tabrecta 400 mg orally twice daily until disease progression or unacceptable toxicity. The major efficacy outcome measure was overall response rate (ORR) as determined by a Blinded Independent Review Committee (BIRC) according to RECIST 1.1. An additional efficacy outcome measure was duration of response (DOR) by BIRC.
The efficacy population included 28 treatment-naïve patients and 69 previously treated patients. The median age was 71 years (range: 49 to 90 years); 60% female; 75% White; 24% had Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) 0 and 75% had ECOG PS 1; 60% never smoked; 80% had adenocarcinoma; and 12% had CNS metastases. Amongst previously treated patients, 88% received prior platinum-based chemotherapy.
Efficacy results are presented in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Pharmacokinetics:
Click on icon to see table/diagram/image
Pharmacokinetics: Capmatinib exposure (AUC0-12h and Cmax) increased approximately proportionally over a dose range of 200 mg (0.5 times the recommended dosage) to 400 mg. Capmatinib reached steady-state by day 3 following twice daily dosing, with a mean (% coefficient of variation [%CV]) accumulation ratio of 1.5 (41%).
Absorption: After administration of Tabrecta 400 mg orally in patients with cancer, capmatinib peak plasma concentrations (Cmax) were reached in approximately 1 to 2 hours (Tmax). The absorption of capmatinib after oral administration is estimated to be greater than 70%.
Effect of Food: A high-fat meal (containing approximately 1000 calories and 50% fat) in healthy subjects increased capmatinib AUC0-INF by 46% with no change in Cmax compared to under fasted conditions. A low-fat meal (containing approximately 300 calories and 20% fat) in healthy subjects had no clinically meaningful effect on capmatinib exposure. When capmatinib was administered at 400 mg orally twice daily in cancer patients, exposure (AUC0-12h) was similar after administration of capmatinib with food and under fasted conditions.
Distribution: Capmatinib plasma protein binding is 96%, independent of capmatinib concentration. The apparent mean volume of distribution at steady-state is 164 L.
The blood-to-plasma ratio was 1.5, but decreased at higher concentrations to 0.9.
Elimination: The effective elimination half-life of capmatinib is 6.5 hours. The mean (%CV) steady-state apparent clearance of capmatinib is 24 L/hr (82%).
Metabolism: Capmatinib is primarily metabolized by CYP3A4 and aldehyde oxidase.
Excretion: Following a single oral administration of radio-labeled capmatinib to healthy subjects, 78% of the total radioactivity was recovered in feces with 42% as unchanged and 22% was recovered in urine with negligible as unchanged.
Specific Populations: No clinically significant effects on the pharmacokinetic parameters of capmatinib were identified for the following covariates assessed: age (26 to 90 years), sex, race (White, Asian, Native American, Black, unknown), body weight (35 to 131 kg), mild to moderate renal impairment (baseline CLcr 30 to 89 mL/min by Cockcroft-Gault) and mild, moderate or severe hepatic impairment (Child-Pugh classification). The effect of severe renal impairment (baseline CLcr 15 to 29 mL/min) on capmatinib pharmacokinetics has not been studied.
Drug Interaction Studies: Clinical Studies and Model-Informed Approaches: Strong CYP3A Inhibitors: Coadministration with itraconazole (a strong CYP3A inhibitor) increased capmatinib AUC0-INF by 42% with no change in capmatinib Cmax.
Strong CYP3A Inducers: Coadministration with rifampicin (a strong CYP3A inducer) decreased capmatinib AUC0-INF by 67% and decreased Cmax by 56%.
Moderate CYP3A Inducers: Coadministration with efavirenz (a moderate CYP3A inducer) was predicted to decrease capmatinib AUC0-12h by 44% and decrease Cmax by 34%.
Proton Pump Inhibitors: Coadministration with rabeprazole (a proton pump inhibitor) decreased capmatinib AUC0-INF by 25% and decreased Cmax by 38%.
Substrates of CYP Enzymes: Coadministration of capmatinib increased caffeine (a CYP1A2 substrate) AUC0-INF by 134% with no change in its Cmax. Coadministration of capmatinib had no clinically meaningful effect on exposure of midazolam (a CYP3A substrate).
P-gp Substrates: Coadministration of capmatinib increased digoxin (a P-gp substrate) AUC0-INF by 47% and increased Cmax by 74%.
BCRP Substrates: Coadministration of capmatinib increased rosuvastatin (a BCRP substrate) AUC0-INF by 108% and increased Cmax by 204%.
In Vitro Studies: Transporter Systems: Capmatinib is a substrate of P-gp, but not a substrate of BCRP or MRP2. Capmatinib reversibly inhibits MATE1 and MATE2K, but does not inhibit OATP1B1, OATP1B3, OCT1, OAT1, OAT3, or MRP2.
Toxicology: Preclinical safety data: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenicity studies were not conducted with capmatinib. Capmatinib was not mutagenic in an in vitro bacterial reverse mutation assay and did not cause chromosomal aberrations in an in vitro chromosome aberration assay in human peripheral blood lymphocytes. Capmatinib was not clastogenic in an in vivo bone marrow micronucleus test in rats.
Dedicated fertility studies were not conducted with capmatinib. No effects on male and female reproductive organs occurred in general toxicology studies conducted in rats and monkeys at doses resulting in exposures of up to approximately 3.6 times the human exposure based on AUC at the 400 mg twice daily clinical dose.
Animal Toxicology and/or Pharmacology: In rats, capmatinib administration resulted in vacuolation of white matter of the brain in both 4- and 13-week studies at doses ≥ 2.2 times the human exposure (AUC) at the 400 mg twice daily clinical dose. In some cases, the brain lesions were associated with early death and/or convulsions or tremors. Concentrations of capmatinib in the brain tissue of rats was approximately 9% of the corresponding concentrations in plasma.
In vitro and in vivo assays demonstrated that capmatinib has some potential for photosensitization; however, the no-observed-adverse-effect level for in vivo photosensitization was 30 mg/kg/day (Cmax of 14000 ng/mL), about 2.9 times the human Cmax at the 400 mg twice daily clinical dose.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
