


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of Action: GAZYVA is a recombinant monoclonal humanized and glycoengineered Type II anti-CD20 antibody of the IgG1 isotype. It specifically targets the extracellular loop of the CD20 transmembrane antigen on the surface of non-malignant and malignant pre-B and mature B-lymphocytes, but not on haematopoietic stem cells, pro-B cells, normal plasma cells or other normal tissue. Glycoengineering of the Fc part of GAZYVA results in higher affinity for FcɣRIII receptors on immune effector cells such as natural killer (NK) cells, and macrophages and monocytes as compared to non-glycoengineered antibodies.
In nonclinical studies, GAZYVA induces direct cell death and mediates antibody dependent cellular cytotoxicity (ADCC) and antibody dependent cellular phagocytosis (ADCP) through recruitment of FcɣRIII positive immune effector cells. In addition, GAZYVA mediates low degree of complement dependent cytotoxicity (CDC). In animal models, GAZYVA mediates potent B cell depletion and antitumour efficacy. Compared to Type I CD20 antibodies, GAZYVA, a Type II antibody, is characterized by an enhanced direct cell death induction with a concomitant reduction in CDC. Compared to non-glycoengineered CD20 antibodies, GAZYVA is characterized by enhanced antibody dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP) as a consequence of the glycoengineering. This translates in superior B cell depletion and anti-tumour efficacy in animal models.
Pharmacodynamic Effects: In the pivotal clinical trial BO21004/CLL11, 91% (40 out of 44) of evaluable patients treated with GAZYVA were B cell depleted (defined as CD19+ B-cell counts < 0.07 x 109/L) at the end of treatment period and remained depleted during the first 6 months of follow up. Recovery of B cells was observed within 12 to 18 months of follow up in 35% (14 out of 40) of patients without progressive disease and 13% (5 out of 40) with progressive disease.
Clinical/Efficacy Studies: Chronic Lymphocytic Leukaemia: A Phase III, international, multicentre, open-label, randomized, two-stage, three-arm study (BO21004/CLL11) investigating the safety and efficacy profile of GAZYVA plus chlorambucil compared to rituximab plus chlorambucil or chlorambucil alone was conducted in patients with previously untreated chronic lymphocytic leukaemia with comorbidities.
Prior to enrolment, patients had to have documented CD20+ CLL, and one or both of the following measures of coexisting medical conditions; comorbidity score [total Cumulative Illness Rating Scale (CIRS)] of greater than 6 or reduced renal function as measured by CrCl <70 mL/min. Patients with inadequate liver function (NCICTC Grade 3 liver function tests (AST, ALT >5 x ULN for >2 weeks; Bilirubin >3 x ULN) and renal function (CrCl <30 mL/min) were excluded.
A total of 781 patients were randomized 2:2:1 to receive GAZYVA plus chlorambucil, rituximab plus chlorambucil or chlorambucil alone. Stage 1 compared GAZYVA plus chlorambucil to chlorambucil alone in 356 patients and Stage 2 compared GAZYVA plus chlorambucil to rituximab plus chlorambucil in 663 patients. Efficacy results are summarized in Table 1 and in Figures 1-3.
In the majority of patients, GAZYVA was given intravenously as a 1000 mg initial dose administered on Day 1, Day 8 and Day 15 of the first treatment cycle. In order to reduce the rate of infusion related reactions in patients, an amendment was implemented and 140 patients received the first GAZYVA dose administered over 2 days [Day 1 (100 mg) and Day 2 (900 mg)] [see Dosage & Administration]. For each subsequent treatment cycle (Cycles 2 to 6), patients received GAZYVA 1000 mg on Day 1 only. Chlorambucil was given orally at 0.5 mg/kg body weight on Day 1 and Day 15 of all treatment cycles (1 to 6).
The demographics data and baseline characteristics were well balanced between the treatment groups. The majority of patients enrolled were Caucasian (95%) and male (61%). The median age was 73 years, with 44% being 75 years or older. At baseline, 22% patients had Binet Stage A, 42% had Binet Stage B and 36% had Binet Stage C. The median comorbidity score was 8 and 76% of the patients enrolled had a comorbidity score above 6. The median estimated CrCl was 62 mL/min and 66% of all patients had a CrCl <70 mL/min. Forty-two percent of patients enrolled had both a CrCl <70 ml/min and a comorbidity score of >6. Thirty-four percent of patients were enrolled on comorbidity score alone, and 23% of patients were enrolled with only impaired renal function. The most frequently reported coexisting medical conditions (using a cut off of 30% or higher), in the MedDRA body systems are: Vascular disorders 73%, Cardiac disorders 46%, GI disorders 38%, Metabolism and Nutrition disorders 40%, Renal and Urinary disorders 38%, musculoskeletal and connective tissue disorders 33%.
The primary endpoint of the study was investigator assessed progression-free survival (PFS-INV). In addition, an independent review committee (IRC) assessed all patients for progression and IRC assessed PFS (PFS-IRC) was evaluated.
Key secondary efficacy endpoints were end of treatment response rate, molecular remission at end of treatment (minimal residual disease status) and time to event endpoints (event-free survival, new anti-leukemic therapy). Overall survival for Stage 1 is presented in Figure 2. Overall survival for stage 2 will continue to be followed and is not yet mature. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResults of the PFS subgroup analysis (i.e. sex, age, Binet stages, CrCl, CIRS score, beta2-microglobulin, IGVH status, chromosomal abnormalities, lymphocyte count at baseline) were consistent with the results seen in the overall Intent-to-Treat population. The risk of disease progression or death was reduced in the GAZYVA plus chlorambucil arm (GClb) compared to the rituximab plus chlorambucil arm (RClb) and chlorambucil alone arm (Clb) in all subgroups. The Hazard Ratios ranged from 0.08 to 0.42 for GClb vs Clb and 0.28 to 0.71 for GClb vs RClb. (See Figures 1, 2 and 3.)
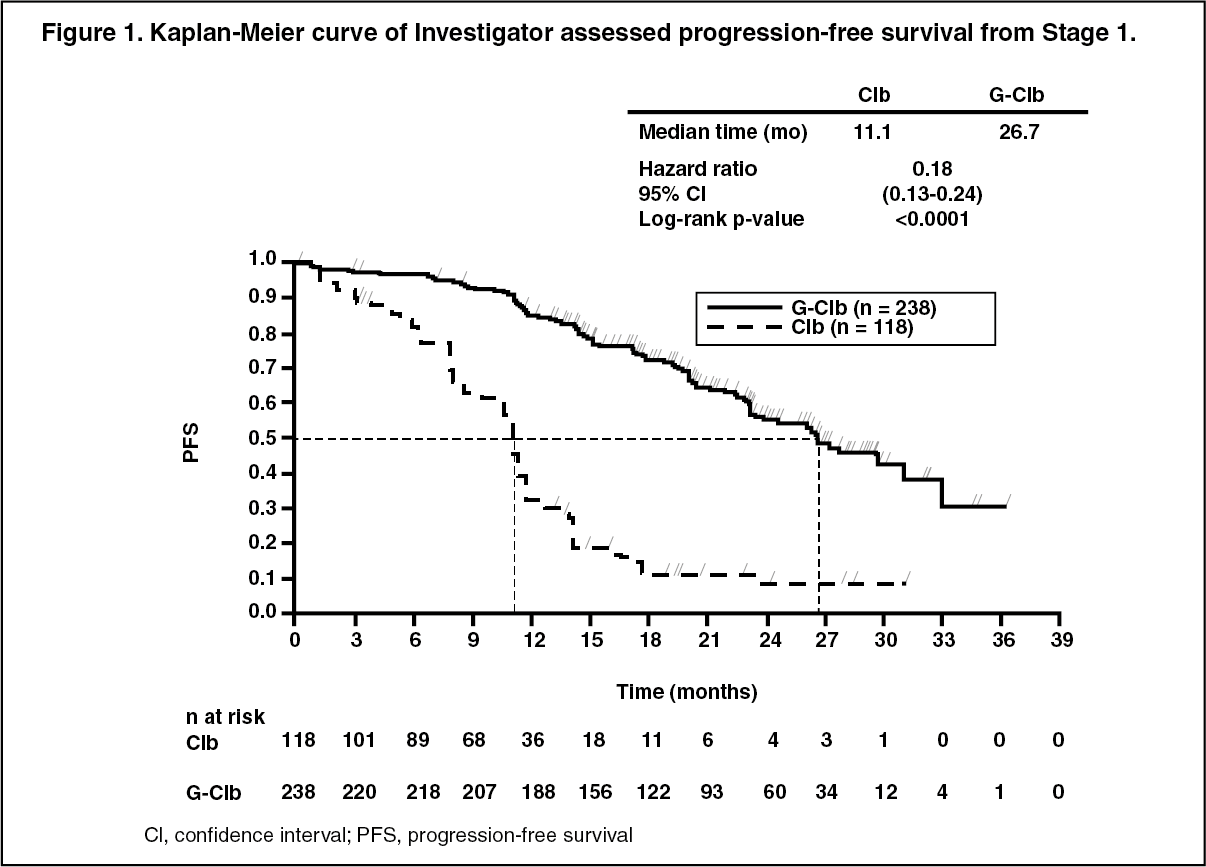 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image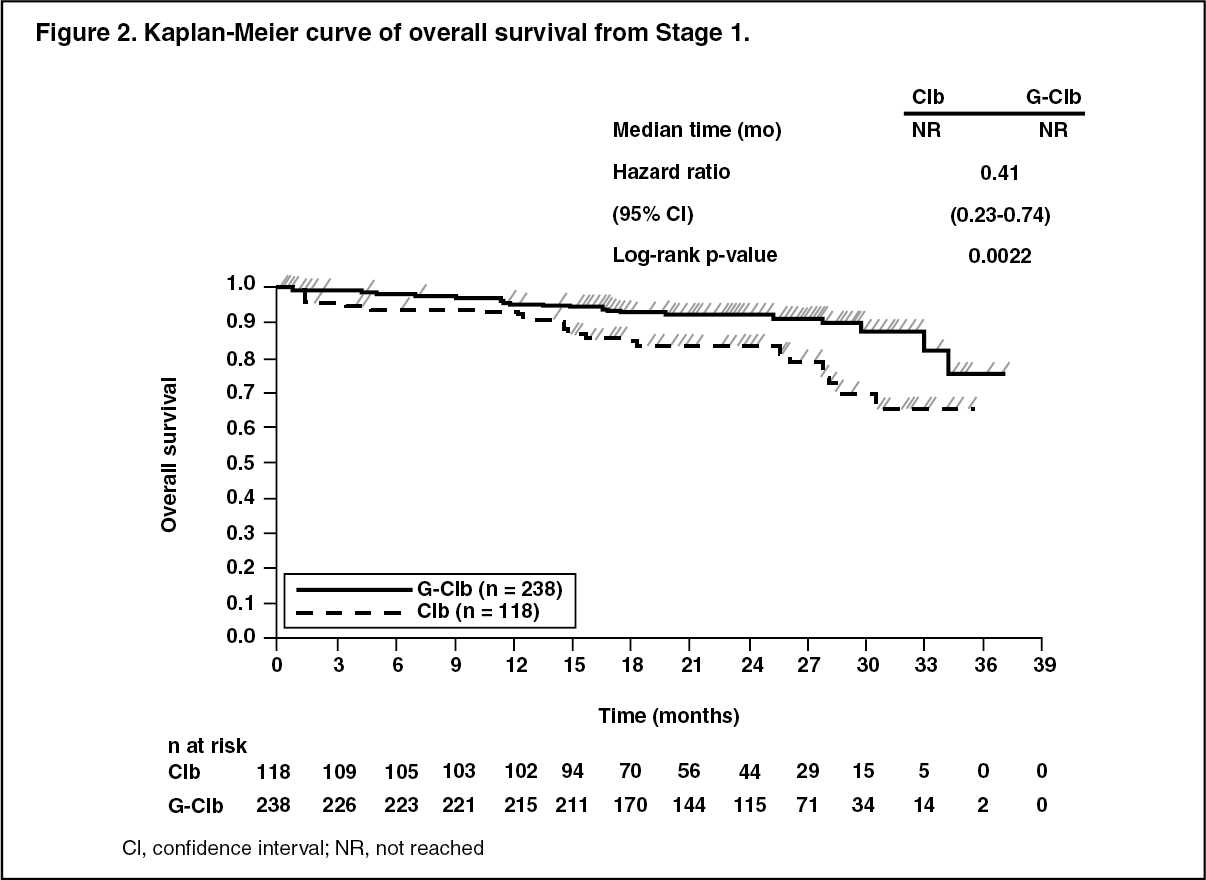 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image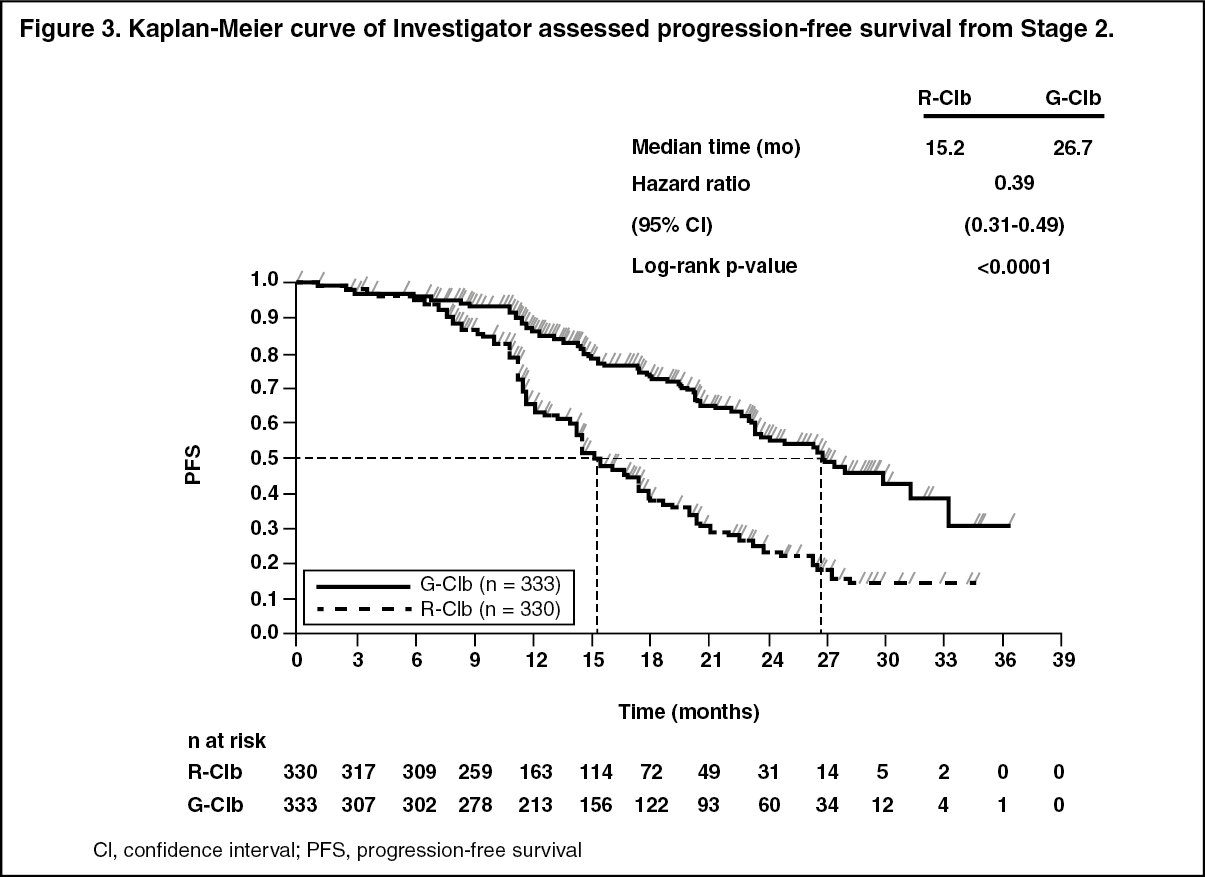 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient Reported Outcomes: In the QLQC30 and QLQ-CLL-16 questionnaires conducted during the treatment period, no substantial difference in any of the subscales was observed. Data during follow up, especially for the chlorambucil alone arm, is limited. However, no notable differences in quality of life during follow up have been identified to date.
Health-related quality of life assessments, specific to fatigue through treatment period, show no statistically significant difference suggesting that the addition of GAZYVA to chlorambucil regimen does not increase the experience of fatigue for patients.
Non-Hodgkin Lymphoma (Follicular Lymphoma): Previously Untreated Follicular Lymphoma: In a multicentre phase III, open-label, randomized study (BO21223/GALLIUM), 1202 previously untreated patients with stage II (bulky)/III/IV follicular lymphoma (FL) were evaluated. Patients were randomized 1:1 to receive either GAZVYA or rituximab in combination with chemotherapy (CHOP, CVP, or bendamustine) followed by GAZYVA or rituximab maintenance in patients who achieved a complete or partial response.
The demographic data and baseline characteristics of the FL population were well balanced [median age was 59 years, the majority of patients were Caucasian (81%), and female (53%)]. Seventy-nine percent had a FLIPI score of ≥2 and 7% had Stage II (bulky), 35% had Stage III and 57% had Stage IV disease. Fifty-seven percent received bendamustine, 33% received CHOP, and 10% received CVP chemotherapy. Forty-four percent had bulky disease (>7 cm), 34% had at least one B-symptom at baseline and 97% had an ECOG performance status of 0-1 at baseline.
GAZYVA (1000 mg) was administered intravenously (as outlined in Dosage & Administration) prior to chemotherapy. Bendamustine was given intravenously on Days 1 and 2 for all treatment cycles (Cycles 1-6) at 90 mg/m2/day when given in combination with GAZYVA. Standard dosing of CHOP and CVP was given. Following Cycles 6-8, when GAZYVA was given in combination with chemotherapy, GAZYVA maintenance therapy was given every 2 months for 2 years for responding patients or until disease progression.
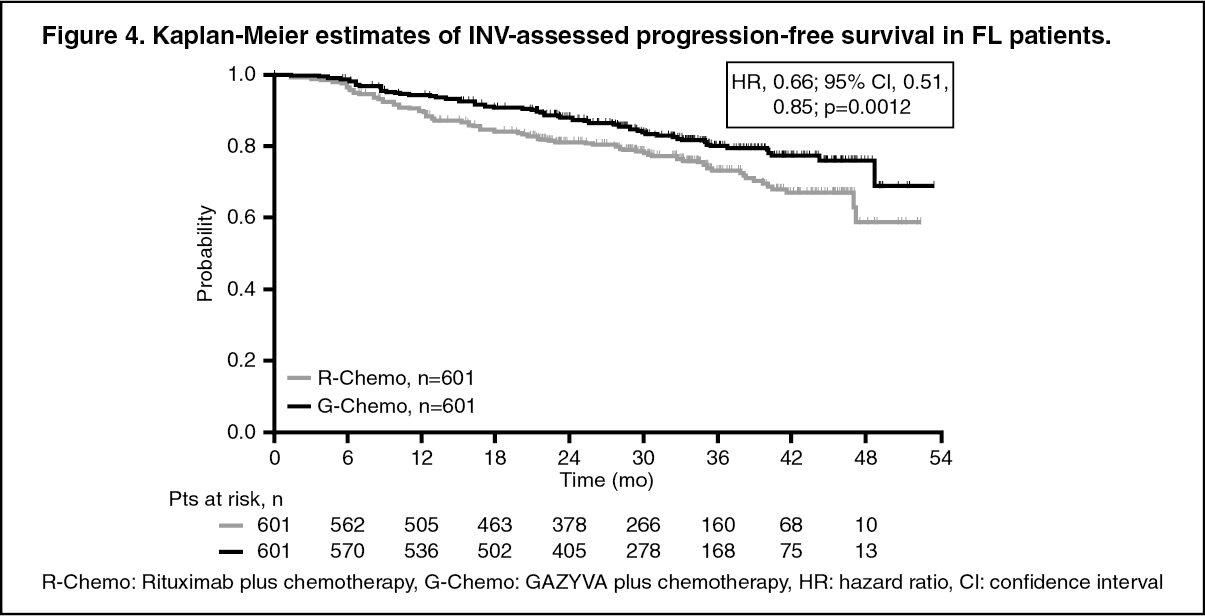
Efficacy results are summarized in Table 2. Kaplan-Meier curves for PFS are shown in Figure 4. (See Table 2.)
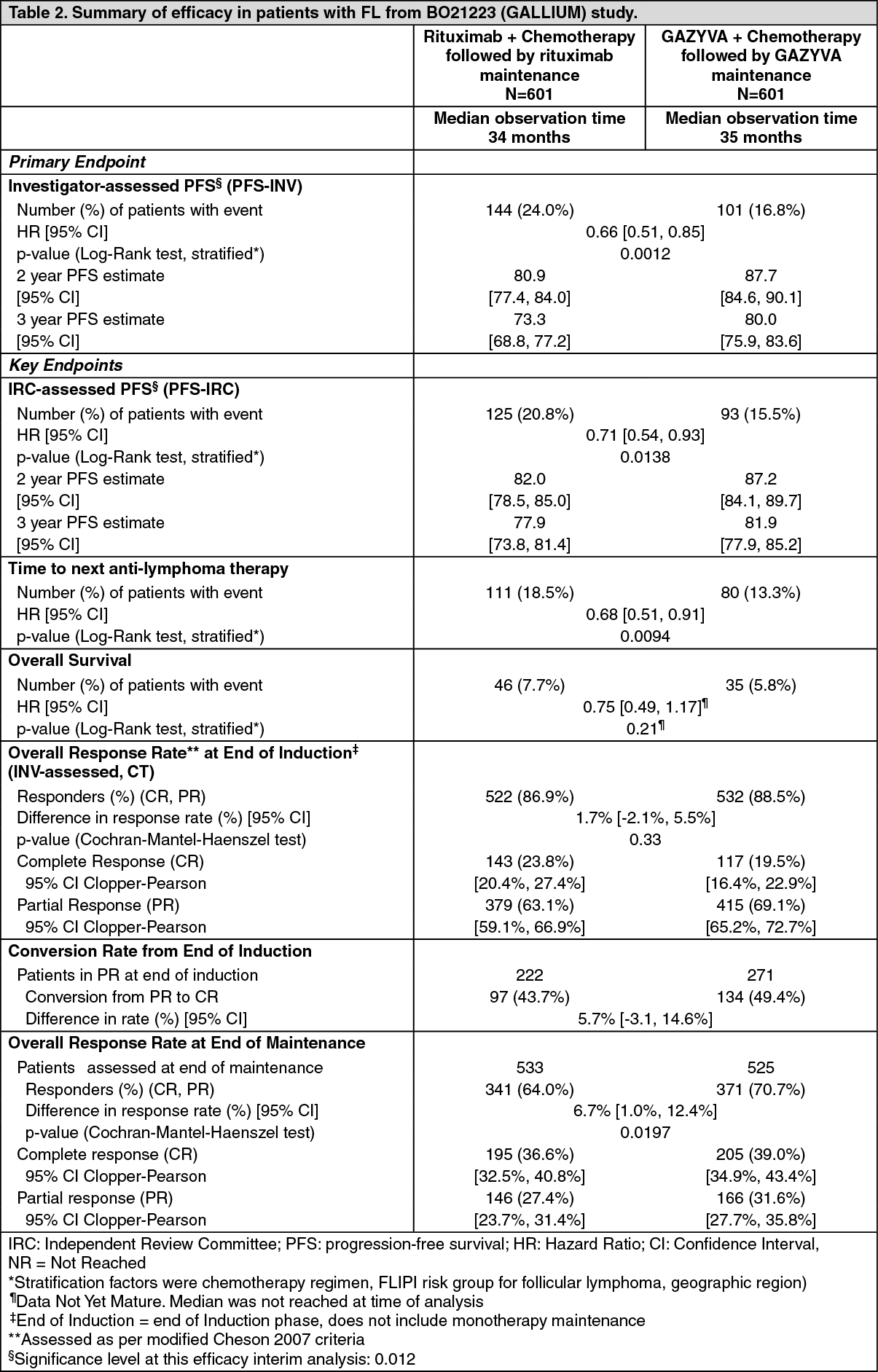 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResponse rates at the end of induction assessed by positron emission tomography (PET) were available for 297/601 patients in the GAZYVA plus chemotherapy arm and 298/601 patients in the rituximab plus chemotherapy arm of the study. Complete response rates at end of induction as assessed by PET were 62.3% in the GAZYVA plus chemotherapy arm and 56.7% in the rituximab plus chemotherapy arm. Overall response rates were similar in the two arms, with a difference of 4.3% in favour of the GAZYVA plus chemotherapy arm (85.9% for G-chemo vs 81.5% for R-chemo). (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResults of subgroup analyses: Results of subgroup analyses were, in general, consistent with the results seen in the FL population, supporting the robustness of the overall result. The subgroups evaluated included IPI, FLIPI, Chemo Regimen, Bulky Disease, B Symptoms at Baseline, Ann Arbor Stage and ECOG at Baseline.
Relapsed/Refractory Follicular Lymphoma: In a phase III, open-label, multicentre, randomized study (GAO4753g/GADOLIN), 396 patients with iNHL who had no response to or who progressed during or up to 6 months after treatment with rituximab or a rituximab-containing regimen were evaluated. Patients were randomized 1:1 to receive either bendamustine (B) alone (n = 202) or GAZYVA in combination with bendamustine (G+B) (n = 194) for 6 cycles, each of 28 days duration. Patients in the G+B arm who did not have disease progression [i.e. patients with a complete response (CR), partial response (PR) or stable disease (SD)] at the end of induction continued receiving GAZYVA maintenance until disease progression or for up to two years (whichever occurred first).
The demographic data and baseline characteristics were well balanced [median age was 63 years; the majority of patients were Caucasian (88%) and male (58%)]. The median time from initial diagnosis was 3 years and the median number of prior therapies was 2 (range 1 to 10); 44% of patients had received 1 prior therapy and 34% of patients had received 2 prior therapies.
GAZYVA was given intravenously as a 1000 mg dose on Days 1, 8 and 15 of Cycle 1, on Day 1 of Cycles 2-6, and in patients who did not have disease progression, every 2 months for up to 2 years or until disease progression. Bendamustine was given intravenously on Days 1 and 2 for all treatment cycles (Cycles 1-6) at 90 mg/m2/day when given in combination with GAZYVA or 120 mg/m2/day when given alone.
The primary analysis demonstrated a statistically significant and clinically meaningful 45% reduction in the risk of disease progression (PD) or death, based on IRC assessment, in patients with iNHL receiving G+B followed by G maintenance vs B alone (stratified log-rank test p-value = 0.0001). IRC-assessed response rates at the end of induction treatment and IRC-assessed best overall response within 12 months of start of treatment were similar in the two treatment arms.
The majority of the patients had follicular lymphoma (FL) (81.1%). Efficacy results from the primary analysis in the FL population are shown in Table 3 and Figures 5 and 6. Of the non-follicular patients, 11.6% had marginal zone lymphoma (MZL) and 7.1% had small lymphocytic lymphoma (SLL). No conclusions could be drawn on efficacy in the MZL and SLL.
At final analysis, the median observation time was 45.9 months (range: 0-100.9 months) for FL patients in the B arm and 57.3 months (range: 0.4-97.6 months) for patients in the G+B arm, representing an additional 25.6 months and 35.2 months of median follow-up in B and G+B arms, respectively, since the primary analysis. Only Investigator (INV) assessed endpoints were reported at final analysis since IRC assessments did not continue. Overall, the efficacy results were consistent with what was observed in the primary analysis. The overall survival (OS) in patients with FL was stable with longer follow-up (see Figure 7); the HR for risk of death was 0.71 (95%CI: 0.51, 0.98). (See Table 3 and Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image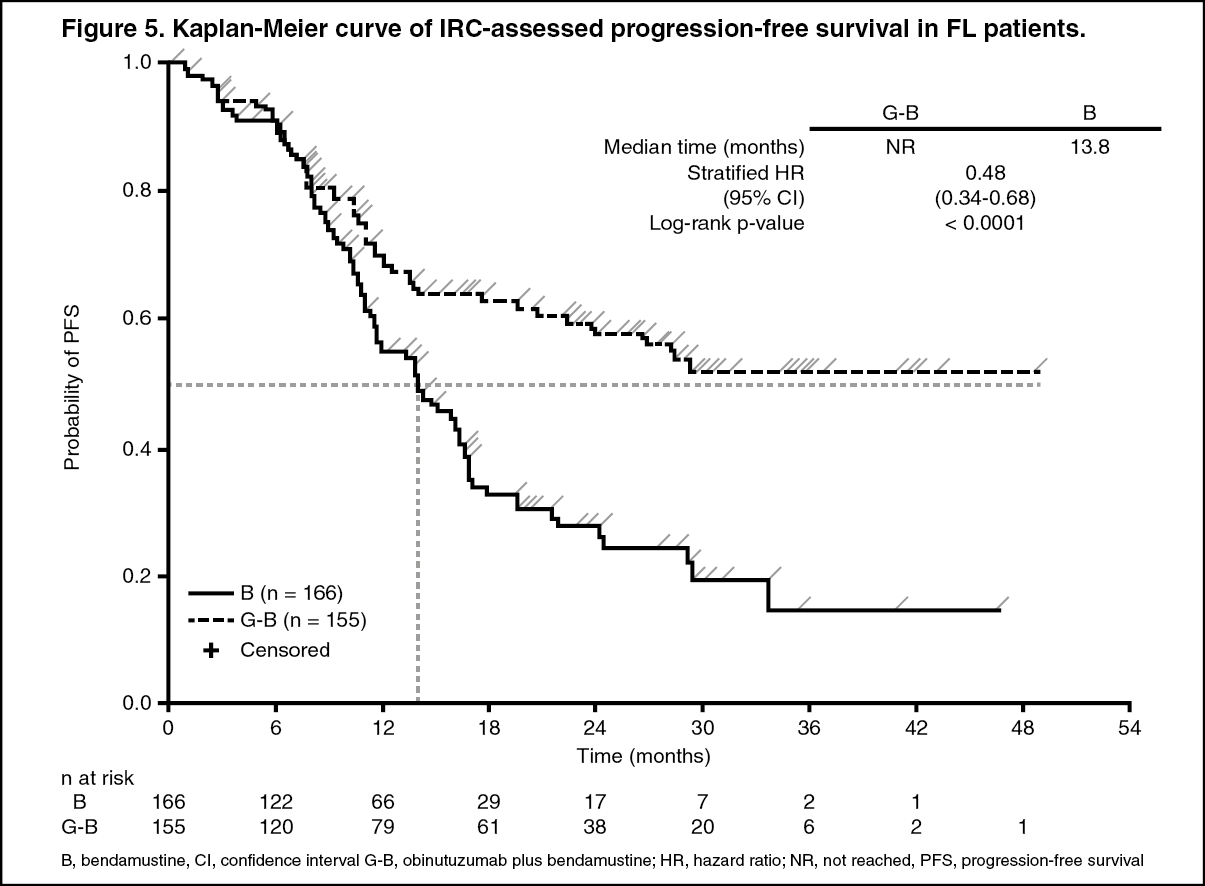 Click on icon to see table/diagram/image
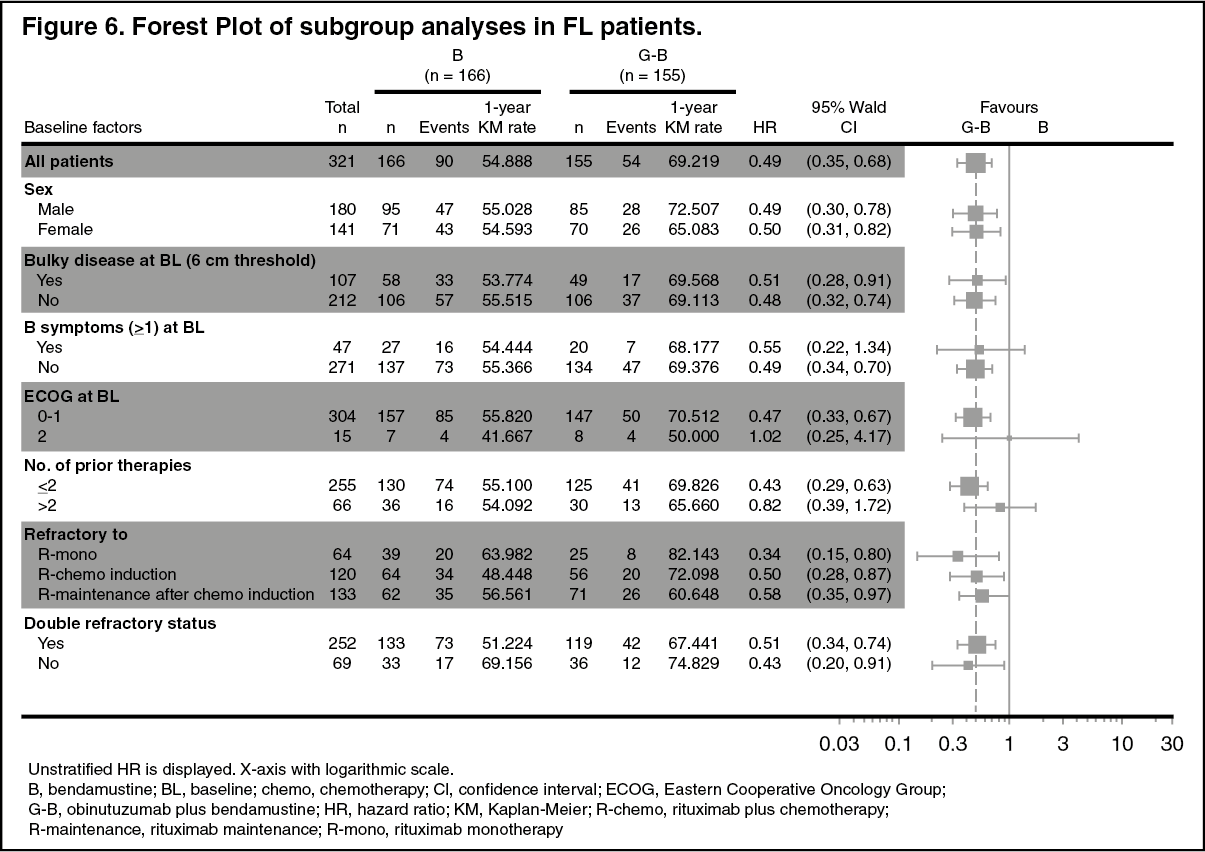
Click on icon to see table/diagram/imageResults of subgroup analyses: Results of subgroup analyses were in general consistent with the results seen in the overall FL population, supporting the robustness of the overall result. (See Figures 6 and 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image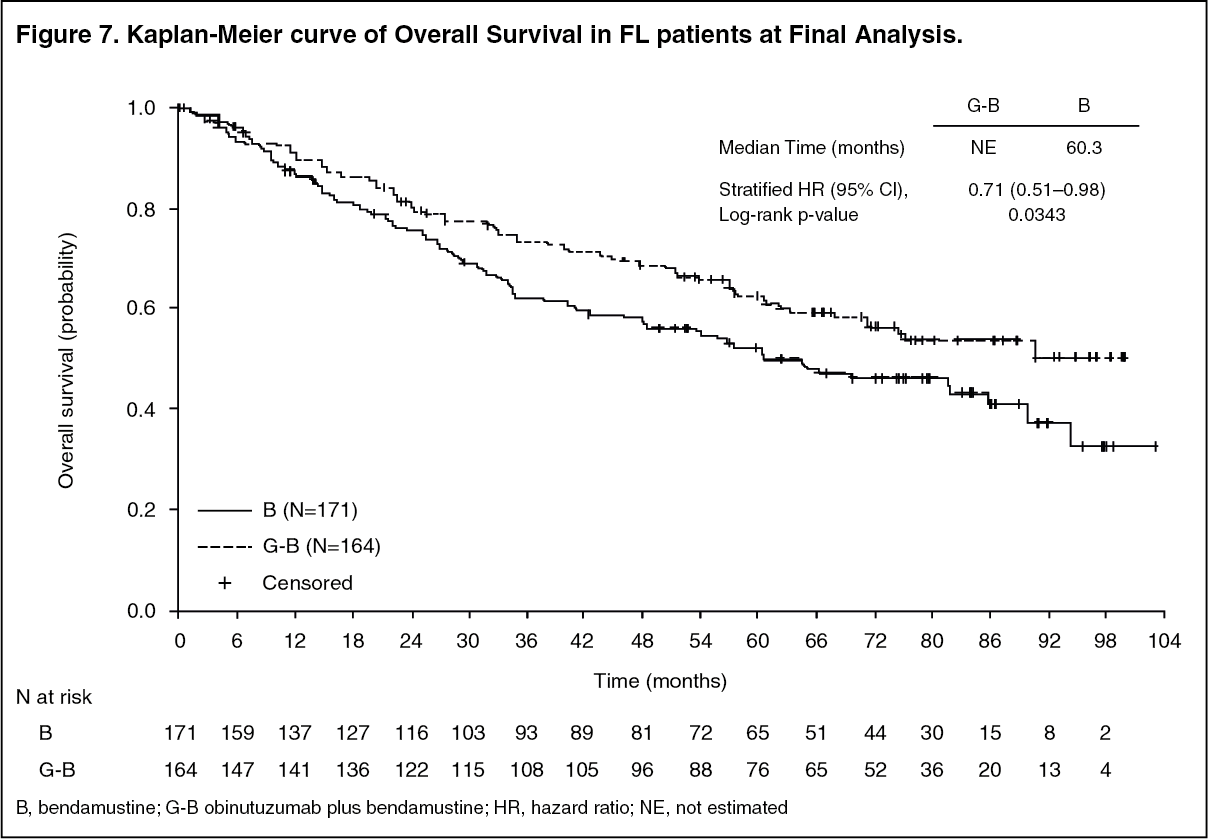 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient Reported Outcomes: Previously Untreated Follicular Lymphoma: Based on the FACT-Lym questionnaire collected during treatment and follow-up periods, both arms experienced clinically meaningful improvements in lymphoma-related symptoms as defined by a ≥ 3 point increase from baseline in the Lymphoma subscale, a ≥ 6 point increase from baseline in the FACT Lym TOI and a ≥ 7 point increase from baseline in the FACT Lym Total score. EQ-5D utility scores were similar at baseline, during treatment and follow-up. No meaningful differences were seen between the arms in HRQOL or health status measures.
Relapsed/Refractory Follicular Lymphoma: Based on the FACT-Lym questionnaire and EQ-5D index scale collected during the treatment and follow-up periods, health-related quality of life was generally maintained in the pivotal study with no meaningful difference between the arms. However, the addition of GAZYVA to bendamustine delayed the time to worsening of quality of life as measured by the FACT-Lym TOI score (HR = 0.83; 95% CI: 0.60, 1.13).
Immunogenicity: Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, assay robustness to quantities of GAZYVA/antibody in circulation, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to GAZYVA with the incidence of antibodies to other products may be misleading.
Patients in the CLL pivotal trial, BO21004/CLL11, were tested at multiple time-points for anti-therapeutic antibodies (ATA) to GAZYVA. In GAZYVA treated patients, 8 out of 140 in the randomized phase and 2 out 6 in the run-in phase tested positive for ATA at 12 months of follow-up. Of these patients, none experienced either anaphylactic or hypersensitivity reactions that were considered related to ATA, nor was clinical response affected.
No post-baseline HAHA (Human Anti-Human Antibody) were observed in patients with iNHL treated in study GAO4753g/GADOLIN. In study BO21223/GALLIUM, 1/565 patient (0.2% of patients with a post-baseline assessment) developed HAHA at induction completion. While the clinical significance of HAHA is not known, a potential correlation between HAHA and clinical course cannot be ruled out.
Pharmacokinetics: A population pharmacokinetic model was developed to analyse the PK data in 469 iNHL, 342 CLL, and 130 DLBCL patients from Phase I, Phase II and Phase III studies who received GAZYVA.
Absorption: GAZYVA is administered intravenously there have been no clinical studies performed with other routes of administration. From the population PK model, after the Cycle 6 Day 1 infusion in CLL patients, the estimated median Cmax value was 465.7 μg/mL and AUC(τ) value was 8,961 μg.d/mL and in iNHL patients the estimated median Cmax value was 539.3 μg/mL and AUC(τ) value was 10,956 μg*d/mL.
Distribution: Following intravenous administration, the volume of distribution of the central compartment (2.72 L), approximates serum volume, which indicates distribution is largely restricted to plasma and interstitial fluid.
Metabolism: The metabolism of GAZYVA has not been directly studied. Antibodies are mostly cleared by catabolism.
Elimination: The clearance of GAZYVA was approximately 0.11 L/day in CLL patients and 0.08 L/day in iNHL patients with a median elimination t ½ of 26.4 days in CLL patients and 36.8 days in iNHL patients. GAZYVA elimination comprises two parallel pathways which describe clearance, a linear clearance pathway and a non-linear clearance pathway which changes as a function of time. During the initial treatment, the non-linear time-varying clearance pathway is dominant and is consequently the major clearance pathway. As treatment continues, the impact of this pathway diminishes and the linear clearance pathway predominates. This is indicative of target mediated drug disposition (TMDD), where the initial abundance of CD20 cells causes a rapid removal of GAZYVA from the circulation. However, once the majority of CD20 cells are bound with GAZYVA, the impact of TMDD on PK is minimized.
Pharmacokinetics in Special Populations: In the population pharmacokinetic analysis, gender was found to be a covariate which explains some of the inter-patient variability, with an 18% greater steady state clearance (CLss) and a 19% greater volume of distribution (V) in males. However, results from the population analysis have shown that the differences in exposure are not significant (with an estimated median AUC and Cmax in CLL patients of 11282 µg*d/mL and 578.9 µg/mL in females and 8451 µg*d/mL and 432.5 µg/mL in males, respectively at Cycle 6, and AUC and Cmax in iNHL patients of 13172 µg*d/mL and 635.7 µg/mL in females and 9769 µg*d/mL and 481.3 µg/mL in males, respectively), indicating that there is no need to dose adjust based on gender.
Geriatric Population: Data obtained in geriatric patients show that pharmacokinetic parameters for GAZYVA are not significantly affected in this population. No significant difference was observed in the pharmacokinetics of GAZYVA among patients <65 years (n=454), patients between 65-75 years (n=317) and patients >75 years (n=190).
Pediatric Population: No studies have been conducted to investigate the pharmacokinetics of GAZYVA in children.
Renal impairment: The population pharmacokinetic analysis of GAZYVA showed that creatinine clearance does not affect pharmacokinetics of GAZYVA. Pharmacokinetics of GAZYVA in patients with mild creatinine clearance (CrCl 50-89 mL/min, n=464) or moderate (CrCl 30 to 49 mL/min, n=106) renal impairment were similar to those in patients with normal renal function (CrCl ≥90 mL/min, n=383). Pharmacokinetic data in patients with severe renal impairment (CrCl 15-29 mL/min) is limited (n=8), therefore no dosage recommendations can be made.
Hepatic impairment: No formal pharmacokinetic study has been conducted and no population PK data was collected in patients with hepatic impairment.
Toxicology: Nonclinical Safety: Carcinogenicity: No carcinogenicity studies have been performed to establish the carcinogenic potential of GAZYVA.
Genotoxicity: No studies have been performed to establish the genotoxic potential of GAZYVA.
Impairment of Fertility: No specific studies in animals have been performed to evaluate the effect of GAZYVA on fertility. No adverse effects on male and female reproductive organs were observed in repeat-dose toxicity studies in cynomolgus monkeys.
Reproductive Toxicity: An enhanced pre- and postnatal development (ePPND) toxicity study was performed on pregnant cynomolgus monkeys. Pregnant animals received weekly intravenous GAZYVA doses [Mean AUC0-168 h at steady state (on Day 139 p.c.) was 125,000 and 250,000 (µg•h)/mL at 25 and 50 mg/kg, respectively. Mean Cmax was 1220 and 2470 µg/mL at 25 and 50 mg/kg, respectively] during gestation (organogenesis period; post-coitum days 20 through delivery). Exposed offspring did not exhibit any teratogenic effects but B-cells were completely depleted on day 28 postpartum. Offspring exposures on day 28 postpartum suggest that GAZYVA can cross the blood-placenta-barrier. Concentrations in infant serum on day 28 postpartum, were in the range of concentrations in maternal serum, whereas concentrations in milk on the same day were very low (less than 0.5% of the corresponding maternal serum levels) suggesting that exposure of infants must have occurred in utero. B-cell counts returned to normal levels, and immunologic function was restored within 6 months postpartum.
Other: In a 26-week cynomolgus monkey study, hypersensitivity reactions were noted and attributed to the foreign recognition of the humanized antibody in cynomolgus monkeys [Cmax and AUC0-168 h at steady state (Day 176) after weekly administration of 5, 25, and 50 mg/kg, were 377, 1530, and 2920 µg/mL and 39,800, 183,000, and 344,000 (µg•h)/mL, respectively]. Findings included acute anaphylactic or anaphylactoid reactions and an increased prevalence of systemic inflammation and infiltrates consistent with immune-complex mediated hypersensitivity reactions, such as arteritis/periarteritis, glomerulonephritis, and serosal/adventitial inflammation. These reactions led to unscheduled termination of 6/36 animals treated with GAZYVA during dosing and recovery phases; these changes were partially reversible. No renal toxicity with a causal relationship to GAZYVA has been observed in humans.