Pharmacotherapeutic group: TNF-alpha Inhibitor.
ATC code: L04AB01.
Pharmacology: Pharmacodynamics: Geriatric use: No specific dosage adjustments of etanercept are recommended based on patient age.
Mechanism of action: Etanercept binds specifically to tumor necrosis factor (TNF) and blocks its interaction with cell surface TNF receptors. TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. It plays an important role in the inflammatory processes of rheumatoid arthritis (RA), polyarticular-course juvenile idiopathic arthritis (JIA), psoriatic arthritis and ankylosing spondylitis and the resulting joint pathology. In addition, TNF plays a role in the inflammatory process of plaque psoriasis. Elevated levels of TNF are found in involved tissues and fluids of patients with RA, JIA, psoriatic arthritis, ankylosing spondylitis (AS) and plaque psoriasis.
Two distinct receptors for TNF (TNFRs), a 55 kilodalton protein (p55) and a 75 kilodalton protein (p75), exist naturally as monomeric molecules on cell surfaces and in soluble forms. Biological activity of TNF is dependent upon binding to either cell surface TNFR.
Etanercept is a dimeric soluble form of the p75 TNF receptor that can bind TNF molecules. Etanercept inhibits binding of both TNF-α and TNF-β (lymphotoxin alpha [LT-α]) to cell surface TNFRs, rendering TNF biologically inactive. In
in-vitro studies, large complexes of etanercept with TNF-α were not detected and cells expressing transmembrane TNF (that binds ENBREL) are not lysed in the presence or absence of complement.
Etanercept can also modulate biological responses that are induced or regulated by TNF, including expression of adhesion molecules responsible for leukocyte migration (i.e., E-selectin and to a lesser extent, intercellular adhesion molecule-1 [ICAM-1]), serum levels of cytokines (e.g., IL-6), and serum levels of matrix metalloproteinase-3 (MMP-3 or stromelysin). Etanercept has been shown to affect several animal models of inflammation, including murine collagen-induced arthritis.
Clinical efficacy: This section presents data from four trials in adults with rheumatoid arthritis, 3 studies in juvenile idiopathic arthritis, 1 study in adults with psoriatic arthritis, 4 studies in adults with ankylosing spondylitis, 1 study in adults with non-radiographic axial spondyloarthritis, 3 studies in adults with plaque psoriasis and 2 studies in pediatric subjects with plaque psoriasis.
Adult patients with rheumatoid arthritis: The efficacy of ENBREL was assessed in a randomized, double-blind, placebo-controlled study. The study evaluated 234 adult patients with active rheumatoid arthritis who had failed therapy with at least one, but no more than four, disease-modifying anti-rheumatic drugs (DMARDs). Doses of 10 mg or 25 mg ENBREL or placebo were administered subcutaneously (SC) twice a week for 6 consecutive months. The results of this controlled trial were expressed in percentage improvement in rheumatoid arthritis using American College of Rheumatology (ACR) response criteria.
ACR 20 and 50 responses were higher in patients treated with ENBREL at 3 and 6 months than in patients treated with placebo (ACR 20: ENBREL 62% and 59%, placebo 23% and 11% at 3 and 6 months, respectively; ACR 50: ENBREL 41% and 40%, placebo 8% and 5% at months 3 and 6, respectively; p<0.01 ENBREL vs. placebo at all timepoints for both ACR 20 and ACR 50 responses).
Approximately 15% of subjects who received ENBREL achieved an ACR 70 response at month 3 and month 6 compared to fewer than 5% of subjects in the placebo arm. Among patients receiving ENBREL, the clinical responses generally appeared within 1 to 2 weeks after initiation of therapy and nearly always occurred by 3 months. A dose response was seen; results with 10 mg were intermediate between placebo and 25 mg. ENBREL was significantly better than placebo in all components of the ACR criteria, as well as other measures of rheumatoid arthritis disease activity not included in the ACR response criteria, such as morning stiffness. A Health Assessment Questionnaire (HAQ), which included disability, vitality, mental health, general health status, and arthritis-associated health status subdomains, was administered every 3 months during the trial. All subdomains of the HAQ were improved in patients treated with ENBREL compared to controls at 3 and 6 months.
After discontinuation of ENBREL, symptoms of arthritis generally returned within a month. Re-introduction of treatment with ENBREL after discontinuations of up to 24 months resulted in the same magnitudes of responses as patients who received ENBREL without interruption of therapy based on results of open-label studies. Continued durable responses have been seen for up to 10 years in open-label extension treatment trials when patients received ENBREL without interruption.
The efficacy of ENBREL was compared to methotrexate in a second randomized, active-controlled study with blinded radiographic evaluations as a primary endpoint in 632 adult patients with active rheumatoid arthritis (<3 years duration) who had never received treatment with methotrexate. Doses of 10 mg or 25 mg ENBREL were administered SC twice a week for up to 24 months. Methotrexate doses were escalated from 7.5 mg/week to a maximum of 20 mg/week over the first 8 weeks of the trial and continued for up to 24 months. Clinical improvement including onset of action within 2 weeks with ENBREL 25 mg was similar to that seen in the previous trials, and was maintained for up to 24 months. At baseline, patients had a moderate degree of disability, with mean HAQ scores of 1.4 to 1.5. Treatment with ENBREL 25 mg resulted in substantial improvement at 12 months, with about 44% of patients achieving a normal HAQ score (less than 0.5). This benefit was maintained in Year 2 of this study.
In this study, structural joint damage was assessed radiographically and expressed as change in Total Sharp Score (TSS) and its components, the erosion score and joint space narrowing score (JSN). Radiographs of hands/wrists and feet were read at baseline and 6, 12, and 24 months. The 10 mg ENBREL dose had consistently less effect on structural damage than the 25 mg dose. ENBREL 25 mg was significantly superior to methotrexate for erosion scores at both 12 and 24 months. The differences in TSS and JSN were not statistically significant between methotrexate and ENBREL 25 mg. The results are shown in the figure as follows. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In another active-controlled, double-blind, randomized study, clinical efficacy, safety, and radiographic progression in RA patients treated with ENBREL alone (25 mg twice weekly), methotrexate alone (7.5 to 20 mg weekly, median dose 20 mg), and of the combination of ENBREL and methotrexate initiated concurrently were compared in 682 adult patients with active rheumatoid arthritis of 6 months to 20 years duration (median 5 years) who had a less than satisfactory response to at least 1 DMARD other than methotrexate.
Patients in the ENBREL in combination with methotrexate therapy group had significantly higher ACR 20, ACR 50, ACR 70 responses and improvement for DAS and HAQ scores at both 24 and 52 weeks than patients in either of the single therapy groups (results shown in table as follows). Significant advantages for ENBREL in combination with methotrexate compared with ENBREL monotherapy and methotrexate monotherapy were also observed after 24 months. (See Table 1.)
Click on icon to see table/diagram/image
Radiographic progression at 12 months was significantly less in the ENBREL group than in the methotrexate group, while the combination was significantly better than either monotherapy at slowing radiographic progression (see figure as follows). (Figure 2.)
Click on icon to see table/diagram/image
Significant advantages for ENBREL in combination with methotrexate compared with ENBREL monotherapy and methotrexate monotherapy were also observed after 24 months. Similarly, the significant advantages for ENBREL monotherapy compared with methotrexate monotherapy were also observed after 24 months.
In an analysis in which all patients who dropped out of the study for any reason were considered to have progressed, the percentage of patients without progression (TSS change ≤0.5) at 24 months was higher in the ENBREL in combination with methotrexate group compared with the ENBREL alone and methotrexate alone groups (62%, 50%, and 36%, respectively; p<0.05). The difference between ENBREL alone and methotrexate alone was also significant (p<0.05). Among patients who completed a full 24 months of therapy in the study, the non-progression rates were 78%, 70%, and 61%, respectively.
The safety and efficacy of 50 mg ENBREL (two 25 mg SC injections) administered once weekly were evaluated in a double-blind, placebo-controlled study of 420 patients with active RA. In this study, 53 patients received placebo; 214 patients received 50 mg ENBREL once weekly, and 153 patients received 25 mg ENBREL twice weekly. The safety and efficacy profiles of the two ENBREL treatment regimens were comparable at week 8 in their effect on signs and symptoms of RA; data at week 16 did not show comparability (non-inferiority) between the two regimens.
Pediatric population with juvenile idiopathic arthritis: The safety and efficacy of etanercept were assessed in a two-part study in 69 children with polyarticular-course juvenile idiopathic arthritis who had a variety of juvenile idiopathic arthritis onset types (polyarthritis, pauciarthritis, systemic-onset). Patients ages 4 to 17 years with moderately to severely active polyarticular course juvenile idiopathic arthritis refractory to or intolerant of methotrexate were enrolled; patients remained on a stable dose of a single non-steroidal anti-inflammatory drug and/or prednisone (≤0.2 mg/kg/day or 10 mg maximum). In part 1, all patients received 0.4 mg/kg (maximum 25 mg per dose) etanercept subcutaneously twice weekly. In part 2, patients with a clinical response at day 90 were randomized to remain on etanercept or receive placebo for four months and assessed for disease flair. Responses were measured using the ACR Pedi 30, defined as ≥30% improvement in at least three of six and ≥30% worsening in no more than one of six JRA core set criteria, including active joint count, limitation of motion, physician and patient/parent global assessments, functional assessment, and ESR. Disease flare was defined as a ≥30% worsening in three of six JRA core set criteria and ≥30% improvement in not more than one of the six JRA core set criteria and a minimum of two active joints.
In part 1 of the study, 51 of 69 (74%) patients demonstrated a clinical response and entered part 2. In part 2, 6 of 25 (24%) patients remaining on etanercept experienced a disease flare compared to 20 of 26 (77%) patients receiving placebo (p=0.007). From the start of part 2, the median time to flare was ≥116 days for patients who received etanercept and 28 days for patients who received placebo. Each component of the JRA core set criteria worsened in the arm that received placebo and remained stable or improved in the arm that continued on etanercept. The data suggested the possibility of a higher flare rate among those patients with a higher baseline ESR. Of patients who demonstrated a clinical response at 90 days and entered part 2 of the study, some of the patients remaining on etanercept continued to improve from month 3 through month 7, while those who received placebo did not improve.
In an open-label, safety extension study, 58 pediatric patients from the previously mentioned study (from the age of 4 years at time of enrolment) continued to receive etanercept for up to 10 years. Rates of serious adverse events and serious infections did not increase with long-term exposure.
In another open-label single-arm study, 60 patients with extended oligoarthritis (15 patients aged 2 to 4, 23 patients aged 5 to 11 and 22 patients aged 12 to 17 years old), 38 patients with enthesitis-related arthritis (12 to 17 years old), and 29 patients with psoriatic arthritis (12 to 17 years old) were treated with etanercept at a dose of 0.8 mg/kg (up to a maximum of 50 mg per dose) administered weekly for 12 weeks. In each of the JIA subtypes, the majority of patients met ACR Pedi 30 criteria and demonstrated clinical improvement in secondary endpoints such as number of tender joints and physician global assessment. The safety profile was consistent with that observed in other JIA studies.
Studies have not been done in patients with juvenile idiopathic arthritis to assess the effects of continued etanercept therapy in patients who do not respond within 3 months of initiating etanercept therapy. Additionally, studies have not been conducted to assess the effects of discontinuing or reducing the recommended dose of etanercept following its long-term use in patients with JIA.
Long-term safety of etanercept monotherapy (n=103), etanercept plus methotrexate (n=294), or methotrexate monotherapy (n=197) were assessed for up to 3 years in a registry of 594 children aged 2 to 18 years with juvenile idiopathic arthritis, 39 of whom were 2 to 3 years of age. Overall, infections were more commonly reported in patients treated with etanercept compared to methotrexate alone (3.8% versus 2%), and the infections associated with etanercept use were of amore severe nature.
Adult patients with psoriatic arthritis: The efficacy of ENBREL was assessed in a randomized, double-blind, placebo-controlled study in 205 patients with psoriatic arthritis. Patients were between 18 and 70 years of age and had active psoriatic arthritis (≥3 swollen joints and ≥3 tender joints) in at least one of the following forms: distal interphalangeal (DIP) involvement; polyarticular arthritis (absence of rheumatoid nodules and presence of psoriasis); arthritis mutilans; asymmetric psoriatic arthritis; or spondylitis-like ankylosis. Patients also had plaque psoriasis with a qualifying target lesion ≥2 cm in diameter. Patients had previously been treated with NSAIDs (86%), DMARDs (80%), and corticosteroids (24%). Patients currently on methotrexate therapy (stable for ≥2 months) could continue at a stable dose of ≤25 mg/week methotrexate. Doses of 25 mg ENBREL (based on dose-finding studies in patients with rheumatoid arthritis) or placebo were administered SC twice a week for 6 months. At the end of the double-blind study, patients could enter a long-term open-label extension study for a total duration of up to 2 years.
Clinical responses were expressed as percentages of patients achieving the ACR 20, 50, and 70 response and percentages with improvement in Psoriatic Arthritis Response Criteria (PsARC). Results are summarized in the table as follows. (See Table 2.)
Click on icon to see table/diagram/image
Among patients with psoriatic arthritis who received ENBREL, the clinical responses were apparent at the time of the first visit (4 weeks) and were maintained through 6 months of therapy. ENBREL was significantly better than placebo in all measures of disease activity (p<0.001), and responses were similar with and without concomitant methotrexate therapy. Quality of life in psoriatic arthritis patients was assessed at every timepoint using the disability index of the HAQ. The disability index score was significantly improved at all timepoints in psoriatic arthritis patients treated with ENBREL, relative to placebo (p<0.001).
Radiographic changes were assessed in the psoriatic arthritis study. Radiographs of hands and wrists were obtained at baseline and months 6, 12, and 24. The modified TSS at 12 months is presented in the table as follows. In an analysis in which all patients who dropped out of the study for any reason were considered to have progressed, the percentage of patients without progression (TSS change ≤0.5) at 12 months was higher in the ENBREL group compared with the placebo group (73% vs. 47%, respectively, p≤0.001). The effect of ENBREL on radiographic progression was maintained in patients who continued on treatment during the second year. The slowing of peripheral joint damage was observed in patients with polyarticular symmetrical joint involvement. (See Table 3.)
Click on icon to see table/diagram/image
ENBREL treatment resulted in improvement in physical function during the double-blind period, and this benefit was maintained during the longer-term exposure of up to 2 years.
There is insufficient evidence of the efficacy of ENBREL in patients with ankylosing spondylitis-like and arthritis mutilans psoriatic arthropathies due to the small number of patients studied.
No study has been performed in patients with psoriatic arthritis using the 50 mg, once-weekly dosing regimen. Evidence of efficacy for the once-weekly dosing regimen in this patient population has been based on data from the study in patients with ankylosing spondylitis.
Adult patients with ankylosing spondylitis: The efficacy of ENBREL in ankylosing spondylitis was assessed in 3 randomized, double-blind studies comparing twice-weekly administration of 25 mg ENBREL with placebo. A total of 401 patients were enrolled, from which 203 were treated with ENBREL. The largest of these trials (n=277) enrolled patients who were between 18 and 70 years of age and had active ankylosing spondylitis defined as visual analog scale (VAS) scores of ≥30 for average of duration and intensity of morning stiffness plus VAS scores of ≥30 for at least 2 of the following 3 parameters: patient global assessment; average of VAS values for nocturnal back pain and total back pain; average of 10 questions on the Bath Ankylosing Spondylitis Functional Index (BASFI). Patients receiving DMARDs, NSAIDs, or corticosteroids could continue them on stable doses. Patients with complete ankylosis of the spine were not included in the study. Doses of 25 mg of ENBREL (based on dose-finding studies in patients with rheumatoid arthritis) or placebo were administered subcutaneously twice a week for 6 months in 138 patients.
The primary measure of efficacy (ASAS 20) was a ≥20% improvement in at least 3 of the 4 Assessment in Ankylosing Spondylitis (ASAS) domains (patient global assessments, back pain, BASFI, and inflammation) and absence of deterioration in the remaining domain. ASAS 50 and 70 responses used the same criteria with a 50% improvement or a 70% improvement, respectively.
Compared to placebo, treatment with ENBREL resulted in significant improvements in the ASAS 20, ASAS 50 and ASAS 70 as early as 2 weeks after the initiation of therapy. (See Table 4.)
Click on icon to see table/diagram/image
Among patients with ankylosing spondylitis who received ENBREL, the clinical responses were apparent at the time of the first visit (2 weeks) and were maintained through 6 months of therapy. Responses were similar in patients who were or were not receiving concomitant therapies at baseline.
Similar results were obtained in the 2 smaller ankylosing spondylitis trials.
In a fourth study, the safety and efficacy of 50 mg ENBREL (two 25 mg SC injections) administered once weekly vs. 25 mg ENBREL administered twice weekly were evaluated in a double-blind, placebo-controlled study of 356 patients with active ankylosing spondylitis. The safety and efficacy profiles of the 50 mg once-weekly and 25 mg twice-weekly regimens were similar.
Adult patients with non-radiographic axial spondyloarthritis: The efficacy of ENBREL in patients with non-radiographic axial spondyloarthritis (nr-AxSpa) was assessed in a randomized, 12-week double-blind, placebo-controlled study. The study evaluated 215 adult patients (modified intent-to-treat population) with active nr-AxSpa (18 to 49 years of age), defined as those patients meeting the ASAS classification criteria of axial spondyloarthritis but did not meet the modified New York criteria for AS. Patients were also required to have an inadequate response to two or more NSAIDs. In the double-blind period, patients received ENBREL 50 mg weekly or placebo for 12 weeks. The primary measure of efficacy (ASAS 40) was a 40% improvement in at least three of the four ASAS domains and absence of deterioration in the remaining domain. MRIs of the sacroiliac joint and spine were obtained to assess inflammation at baseline and at week 12. The double-blind period was followed by an open-label period during which all patients receive ENBREL 50 mg weekly for up to an additional 92 weeks.
Compared to placebo, treatment with ENBREL resulted in statistically significant improvement in the ASAS 40, ASAS 20 and ASAS 5/6. Significant improvement was also observed for the ASAS partial remission and BASDAI 50. Week 12 results are shown in the table as follows. (See Table 5.)
Click on icon to see table/diagram/image
At week 12, there was a statistically significant improvement in the SPARCC (Spondyloarthritis Research Consortium of Canada) score for the sacroiliac joint as measured by MRI for patients receiving ENBREL. Adjusted mean change from baseline was 3.8 for ENBREL treated (n=95) versus 0.8 for placebo treated (n=105) patients (p<0.001).
Health-related quality of life and physical function were assessed using the BASFI (Bath Ankylosing Spondylitis Functional Index), EuroQol 5D and the SF-36 questionnaires. ENBREL showed statistically significantly greater improvement in the BASFI, EQ5D Overall Health State Score and the SF-36 Physical Component Score (PCS) from baseline to week 12 compared to placebo.
Clinical responses among nr-AxSpa patients who received ENBREL were apparent at the time of the first visit (2 weeks) and were maintained through 2 years of therapy. Improvements in health-related quality of life and physical function were also maintained through 2 years of therapy. The 2 year data did not reveal any new safety findings.
Adult patients with plaque psoriasis: The safety and efficacy of ENBREL in patients with plaque psoriasis were assessed in three randomized, double-blind, placebo-controlled studies. The primary efficacy endpoint in all three studies was the proportion of patients in each treatment group who achieved the PASI 75 (i.e., at least a 75% improvement in the Psoriasis Area and Severity Index score from baseline) at 12 weeks.
Study 1 was a Phase 2 study in patients with active, but clinically stable plaque psoriasis involving ≥10% of the body surface area who were ≥18 years old. One hundred and twelve (112) patients were randomized to receive a dose of 25 mg of ENBREL (n=57) or placebo (n=55) twice a week for 24 weeks.
Study 2 evaluated 652 patients with chronic plaque psoriasis, using the same inclusion criteria as study 1, with the addition of a minimum psoriasis area and severity index (PASI) of 10 at screening. ENBREL was administered at doses of 25 mg once a week, 25 mg twice a week or 50 mg twice a week for 6 consecutive months. During the first 12 weeks of the double-blind treatment period, patients received placebo or one of the previously mentioned three ENBREL doses. After 12 weeks of treatment, patients in the placebo group began treatment with blinded ENBREL (25 mg twice a week); patients in the active treatment groups continued to week 24 on the dose to which they were originally randomized.
Study 3 evaluated 583 patients and had the same inclusion criteria as study 2. Patients in this study received a dose of 25 mg or 50 mg ENBREL, or placebo twice a week for 12 weeks, and then all patients received open-label 25 mg ENBREL twice weekly for an additional 24 weeks.
In study 1, the ENBREL-treated group had a significantly higher proportion of patients with a PASI 75 response at week 12 (30%) compared to the placebo-treated group (2%) [p<0.0001]. At 24 weeks, 56% of patients in the ENBREL-treated group had achieved the PASI 75 compared to 5% of placebo-treated patients. Key results of studies 2 and 3 are shown as follows. (See Table 6.)
Click on icon to see table/diagram/image
Among patients with plaque psoriasis who received ENBREL, significant responses relative to placebo were apparent at the time of the first visit (2 weeks) and were maintained through 24 weeks of therapy.
Study 2 also had a drug withdrawal period, during which patients who achieved a PASI improvement of at least 50% at week 24 had treatment stopped. Patients were observed off treatment for the occurrence of rebound (PASI ≥150% of baseline) and for the time to relapse (defined as a loss of at least half of the improvement achieved between baseline and week 24). During the withdrawal period, symptoms of psoriasis gradually returned with a median time to disease relapse of 3 months. No rebound flare of disease and no psoriasis-related serious adverse events were observed. There was some evidence to support a benefit of re-treatment with ENBREL in patients initially responding to treatment.
In study 3, the majority of patients (77%) who were initially randomized to 50 mg twice weekly and had their ENBREL dose decreased at week 12 to 25 mg twice weekly maintained their PASI 75 response through week 36. For patients who received 25 mg twice weekly throughout the study, the PASI 75 response continued to improve between weeks 12 and 36.
In long-term (up to 34 months), open-label studies where ENBREL was given without interruption, clinical responses were sustained and safety was comparable to shorter-term studies.
Pediatric patients with plaque psoriasis: The efficacy of ENBREL was assessed in a randomized, double-blind, placebo-controlled study in 211 pediatric patients aged 4 to 17 years with moderate to severe plaque psoriasis (as defined by a sPGA score ≥3, involving ≥10% of the BSA, and PASI ≥12). Eligible patients had a history of receiving phototherapy or systemic therapy, or were inadequately controlled on topical therapy.
Patients received ENBREL 0.8 mg/kg (up to 50 mg) or placebo once weekly for 12 weeks. At week 12, more patients randomized to ENBREL had positive efficacy responses (e.g., PASI 75) than those randomized to placebo. (See Table 7.)
Click on icon to see table/diagram/image
After the 12-week double-blind treatment period, all patients who entered the open-label period received ENBREL 0.8 mg/kg (up to 50 mg) once weekly for additional 24 weeks. Responses observed during the open-label period were similar to those observed in the double-blind period.
During a randomized withdrawal period, significantly more patients re-randomized to placebo experienced disease relapse (loss of PASI 75 response) compared with patients re-randomized to ENBREL. With continued therapy, responses were maintained up to 48 weeks.
The long-term safety and effectiveness of ENBREL 0.8 mg/kg (up to 50 mg) once weekly was assessed in an open-label extension study of 181 pediatric subjects with plaque psoriasis for up to 2 years beyond the 48 week study discussed previously. Long-term experience with ENBREL was generally comparable to the original 48-week study and did not reveal any new safety findings.
Pharmacokinetics: Absorption: Etanercept is slowly absorbed from the site of SC injection, reaching maximum concentration approximately 48 hours after a single dose. The absolute bioavailability is 76%.
Distribution: After a single SC dose of 25 mg etanercept, the average maximum serum concentration observed in healthy volunteers was 1.65 ± 0.66 μg/mL, and the area under the curve was 235 ± 96.6 μg·hr/mL. Dose proportionality has not been formally evaluated, but there is no apparent saturation of clearance across the dosing range.
The volume of distribution at steady-state after subcutaneous administration is 13.9 ± 9.4 L.
After continued dosing of RA patients (n=25) with etanercept for 6 months with 25 mg twice weekly, the median observed level was 3.0 μg/mL (range 1.7 to 5.6 μg/mL). Based on the available data, individual patients may undergo a two- to five-fold increase in serum levels with repeated dosing.
Elimination: Etanercept is cleared slowly from the body. The half-life is approximately 80 hours.
The clearance is approximately 175 ± 116 mL/hr in patients with rheumatoid arthritis and 131 ± 81 mL/hr in healthy volunteers.
Radioactivity is eliminated in urine after administration of radiolabeled etanercept to patients and volunteers.
Renal impairment or hepatic impairment: Although there is elimination of radioactivity in urine after administration of radiolabeled etanercept to patients and volunteers, increased etanercept concentrations were not observed in patients with acute renal or hepatic failure. The presence of renal or hepatic impairment should not require a change in dosage.
Gender: There is no apparent pharmacokinetic difference between men and women.
Concentration-effect relationship: Steady-state serum concentrations of 1 to 2 mg/L of etanercept are associated with optimal effect and are obtained with doses of 25 mg twice weekly. In an open-label, single-dose, two-treatment, crossover study in 28 healthy volunteers, etanercept, administered as a single 50 mg/mL injection, was found to be bioequivalent to two simultaneous injections of 25 mg/mL.
Toxicology: PRECLINICAL SAFETY DATA: Carcinogenicity, Mutagenicity, and Impairment of Fertility: Long-term animal studies have not been conducted to evaluate the carcinogenic potential of etanercept or its effect on fertility. Long-term animal studies are not feasible because animals can develop antibodies to etanercept, which is a human protein. Mutagenesis studies were conducted
in vitro and in
vivo, and no evidence of mutagenic activity was observed.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image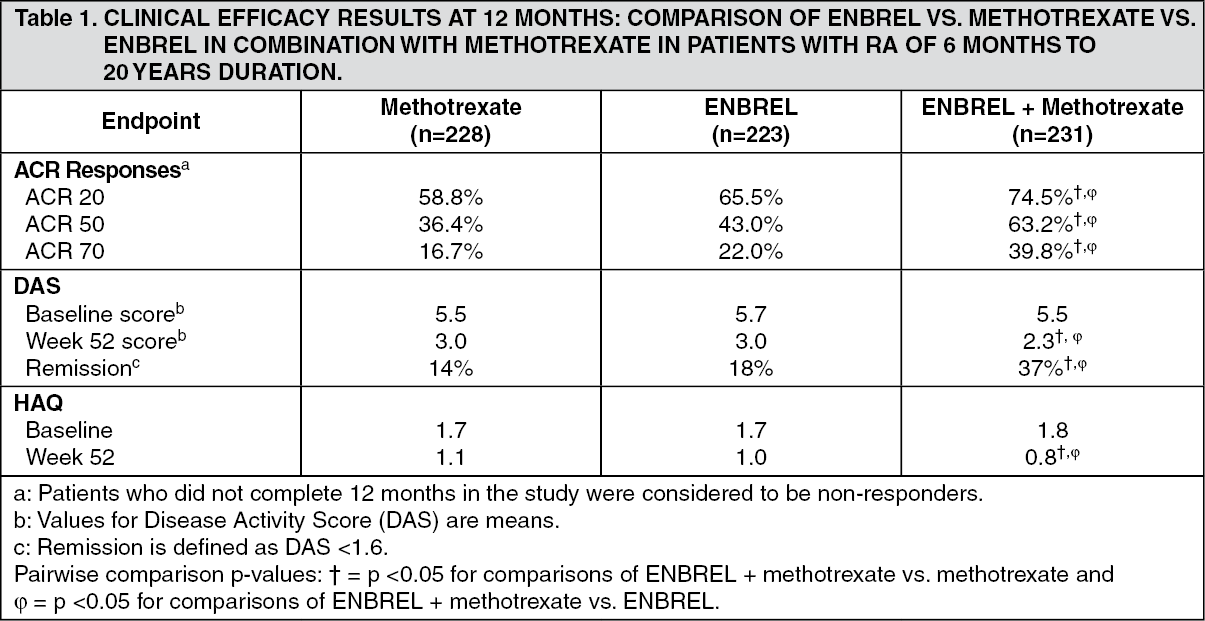 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
