PHARMACOLOGY: Mechanism of Action: The mechanism of action of lemborexant in the treatment of insomnia is presumed to be through antagonism of orexin receptors. The orexin neuropeptide signaling system plays a role in wakefulness. Blocking the binding of wake-promoting neuropeptides orexin A and orexin B to receptors OX1R and OX2R is thought to suppress wake drive.
Pharmacodynamics: Lemborexant binds to orexin receptors OX1R and OX2R and acts as a competitive antagonist (IC
50 values of 6.1 nM and 2.6 nM, respectively). A major metabolite of lemborexant, M10, binds with comparable affinity as the parent drug to orexin receptors OX1R and OX2R (IC
50 values of 4.2 nM and 2.9 nM), respectively.
Cardiac Electrophysiology: In a concentration-QTcF analysis using the data from two randomized, double-blind, placebo-controlled, multiple ascending dose studies in healthy subjects, lemborexant does not prolong the QTcF interval to any clinically relevant extent at a dose 5 times the maximum recommended dose.
Drug Interactions: Lemborexant co-administered with alcohol produced a numerically greater negative impact on postural stability and memory as compared with alcohol alone at approximately 2 hours post-dose [see Interactions].
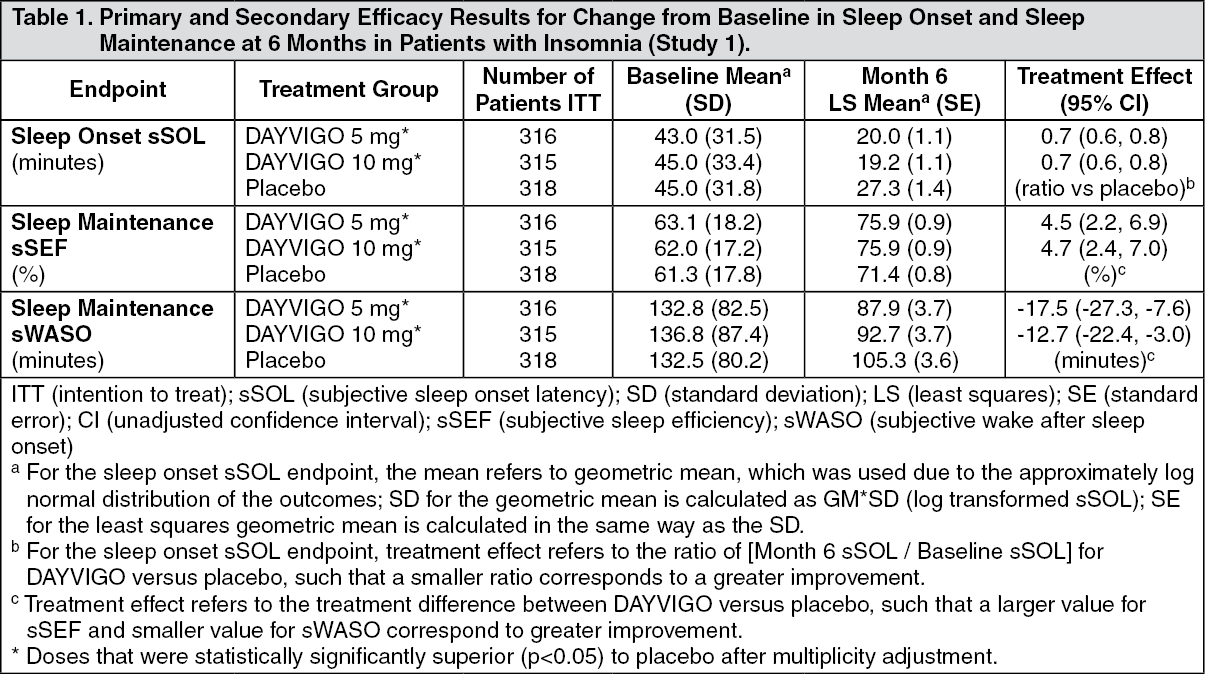
CLINICAL STUDIES: Controlled Clinical Studies: DAYVIGO was evaluated in two clinical trials in patients with insomnia characterized by difficulties with sleep onset and/or sleep maintenance (Study 1, NCT02952820 and Study 2, NCT02783729).
Study 1 was a 6-month, randomized, double-blind, placebo-controlled, multi-center trial in adult patients age 18 or older who met DSM-5 criteria for insomnia disorder. Patients were randomized to placebo (n=325), DAYVIGO 5 mg (n=323), or DAYVIGO 10 mg (n=323) once nightly. The primary efficacy endpoint was the mean change from baseline to end of treatment at 6 months for log-transformed patient-reported (subjective) sleep onset latency (sSOL), defined as the estimated minutes from the time that the patient attempted to sleep until sleep onset. Pre-specified secondary efficacy endpoints for sleep maintenance were change from baseline to end of treatment at 6 months for patient-reported sleep efficiency (sSEF) and wake after sleep onset (sWASO). sSEF is defined as the proportion of time spent asleep per time in bed. sWASO is defined as the minutes of wake from the onset of sleep until wake time. The primary and pre-specified secondary efficacy endpoints were measured by sleep diary.
The demographic characteristics of patients in Study 1 were similar across the treatment arms. Patients had a median age of 55 years (range 18 to 88) and were 68% female, 72% White, 8% Black or African American, 17% Japanese, and 3.5% other; 28% were elderly (≥65 years).
Examination of subgroups by age, race, and sex did not suggest differences in response to DAYVIGO. In Study 1, DAYVIGO 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure, sSOL, compared to placebo (Table 1). DAYVIGO 5 mg and 10 mg also showed statistically significant superiority in sSEF and sWASO. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Study 2 was a 1-month, randomized, double-blind, placebo- and active-controlled, multi-center, parallel-group clinical trial in adult female patients age 55 and older and male patients 65 years and older who met DSM-5 criteria for insomnia disorder. Patients were randomized to placebo (n=208), DAYVIGO 5 mg (n=266) or 10 mg (n=269), or active comparator (n=263) once nightly.
The primary efficacy endpoint was the mean change in log-transformed latency to persistent sleep (LPS) from baseline to end of treatment (Days 29/30), as measured by overnight polysomnography (PSG) monitoring. LPS was defined as the number of minutes from lights off to the first 10 consecutive minutes of non-wakefulness. The pre-specified secondary efficacy endpoints in Study 2 were the mean change from baseline to end of treatment (Days 29/30) in sleep efficiency (SEF) and wake after sleep onset (WASO) measured by PSG.
The demographic and baseline characteristics of patients in Study 2 were similar across the treatment arms. Patients had a median age of 63 years (range 55 to 88) and were 86% female, 72% White, 25% Black or African American, and 2% other; 45% were elderly (≥65 years).
In Study 2, DAYVIGO 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure, LPS, compared to placebo (Table 2). DAYVIGO 5 mg and 10 mg demonstrated statistically significant improvement in SEF and WASO compared to placebo. (See Table 2.)
Click on icon to see table/diagram/image
The effects of DAYVIGO at the beginning of treatment were generally consistent with later timepoints.
Special Safety Studies: Middle of the Night Safety: The effect of DAYVIGO on middle of the night safety was evaluated in a randomized, placebo- and active-controlled trial in healthy female subjects ≥55 years or male subjects ≥65 years. Postural stability, the ability to awaken in response to a sound stimulus, and attention and memory were assessed following a scheduled awakening 4 hours after the start of the 8-hour time in bed. Postural stability was measured by assessing body sway using an ataxia meter. Nighttime dosing of DAYVIGO 5 mg and 10 mg resulted in impairment of balance (measured by body sway area) at 4 hours as compared to placebo.
The ability to awaken to sound in the middle of the night was assessed using an audiometer that delivered 1000 Hz tones up to 105 dB. There were no meaningful differences between DAYVIGO (5 mg or 10 mg) and placebo on ability to awaken to sound.
A computerized performance assessment battery was administered to assess attention and memory after middle of the night awakening (4 hours postdose) in subjects receiving DAYVIGO 5 mg or 10 mg. DAYVIGO was associated with dose-dependent worsening on measures of attention and memory as compared to placebo.
Patients should be cautioned about the potential for middle of the night postural instability, as well as attention and memory impairment.
Effects on Next-day Postural Stability and Memory: The effects of DAYVIGO on next day postural stability and memory were evaluated in two randomized, placebo- and active-controlled trials in healthy subjects and insomnia patients age 55 and older.
There were no meaningful differences between DAYVIGO (5 mg or 10 mg) and placebo on next-day postural stability or memory compared to placebo.
Effects on Driving: A randomized, double-blind, placebo- and active-controlled, four-period crossover study evaluated the effects of nighttime administration of DAYVIGO on next-morning driving performance approximately 9 hours after dosing in 24 healthy elderly subjects (≥65 years, median age 67 years; 14 men, 10 women) and 24 adult subjects (median age 49 years; 12 men, 12 women). The primary driving performance outcome measure was change in Standard Deviation of Lateral Position (SDLP). Testing was conducted after one night (a single dose) and after eight consecutive nights of treatment with DAYVIGO. Although DAYVIGO at doses of 5 mg and 10 mg did not cause statistically significant impairment in next-morning driving performance in adult or elderly subjects (compared with placebo), driving ability was impaired in some subjects taking 10 mg DAYVIGO.
Patients using the 10 mg dose should be cautioned about the potential for next-morning driving impairment because there is individual variation in sensitivity to DAYVIGO.
Rebound Insomnia: Rebound insomnia was assessed by comparing sleep diary-recorded sSOL and sWASO from the screening period to the two weeks following treatment discontinuation in both Studies 1 and 2. Analyses of group means and the proportion of patients with rebound insomnia suggest that DAYVIGO was not associated with rebound insomnia following treatment discontinuation.
Withdrawal Effects: In 12-month and 1-month controlled safety and efficacy trials (Studies 1 and 2, respectively), withdrawal effects were assessed by the Tyrer Benzodiazepine Withdrawal Symptom Questionnaire following discontinuation from study drug in patients who received DAYVIGO 5 mg or 10 mg. There was no evidence of withdrawal effects following DAYVIGO discontinuation at either dose.
Pharmacokinetics: Following single doses of lemborexant 2.5 to 75 mg, geometric mean C
max and AUC
0-24h increased slightly less than in proportion to dose. The extent of accumulation of lemborexant at steady-state is 1.5- to 3-fold across this dose range.
Absorption: The time to peak concentration (t
max) of lemborexant is approximately 1 to 3 hours.
Effect of Food: Lemborexant C
max decreased by 23%, AUC
0-inf increased by 18%, and t
max was delayed by 2 hours following administration of a high-fat and high-calorie meal (containing approximately 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively).
Distribution: The volume of distribution of lemborexant is 1970 L. Protein binding of lemborexant is approximately 94%
in vitro. The blood to plasma concentration ratio of lemborexant is 0.65.
Elimination: Metabolism: Lemborexant is primarily metabolized by CYP3A4, and to a lesser extent by CYP3A5. The major circulating metabolite is M10.
Excretion: Following administration of an oral dose, 57.4% of the dose was recovered in the feces and 29.1% in the urine (<1% as unchanged). The effective half-life for lemborexant 5 mg and 10 mg is 17 and 19 hours, respectively.
Specific Populations: No clinically significant differences in the pharmacokinetics of lemborexant were observed based on age, sex, race/ethnicity, or body mass index. No studies have been conducted to investigate the pharmacokinetics of lemborexant in pediatric patients. Exposures of lemborexant in patients with hepatic and renal impairment are summarized in Figure 1. (See Figure 1.)
Click on icon to see table/diagram/image
Drug Interaction Studies: The effects of other drugs on the exposures of lemborexant are summarized in Figure 2. The effects of lemborexant on the exposures of other drugs are summarized in Figure 3.
Physiologically-based pharmacokinetic (PBPK) modeling predicted that concomitant use of weak CYP3A inhibitors increased lemborexant exposure by less than 2-fold. Based on these results, drug interactions between lemborexant and strong CYP3A inducers, strong CYP3A inhibitors, moderate CYP3A inhibitors, and CYP2B6 substrates are clinically significant. (See Figures 2 and 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In Vitro Studies: In vitro metabolism studies demonstrated that lemborexant and M10 have the potential to induce CYP3A and the weak potential to inhibit CYP3A and induce CYP2B6. Lemborexant and M10 do not inhibit other CYP isoforms (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2A6, CYP2C19, and CYP2E1) or transporters (P-gp, BCRP, BSEP, OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, MATE1, and MATE2-K). Lemborexant is a potential poor substrate of P-gp, but M10 is a substrate of P-gp. Lemborexant and M10 are not substrates of BCRP, OATP1B1, or OATP1B3.
Toxicology: Preclinical safety data: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenesis: Lemborexant did not increase the incidence of tumors in rats treated for 2 years at oral doses of 30, 100, and 300 mg/kg/day (males) and 10, 30, and 100 mg/kg/day (females), which are >80 times the MRHD based on AUC. Lemborexant did not increase the incidence of tumors in Tg ras H2 mice treated for 26 weeks at oral doses of 50, 150, and 500 mg/kg/day.
Mutagenesis: Lemborexant was not mutagenic in the
in vitro bacterial reverse mutation (Ames) assay or in the
in vitro mouse lymphoma thymidine kinase assay, and was not clastogenic in the
in vivo rat micronucleus assay.
Impairment of Fertility: Lemborexant was orally administered to female rats at doses of 30, 100, or 1000 mg/kg/day prior to and throughout mating and continuing to gestation Day 6. These doses are approximately 12 to >500 times the MRHD based on AUC. Irregular estrous cycles and decreased pregnancy rate were observed at 60 times the MRHD based on AUC, and decreased numbers of corpora lutea, implantations, and live embryos were observed at >500 times the MRHD based on AUC. The exposure at the NOAEL of 30 mg/kg/day is approximately 12 times the MRHD based on AUC. Lemborexant did not affect fertility when orally administered to male rats at doses of 30, 100, or 1000 mg/kg/day prior to and throughout mating; the highest dose is approximately 138 times the MRHD based on AUC.
Animal Toxicology and/or Pharmacology: Lemborexant administered to mice at oral doses of 10 or 30 mg/kg resulted in behavior characteristic of cataplexy when presented with chocolate. Chocolate is a stimulus that has been demonstrated to increase cataplexy occurrences in narcoleptic mice.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image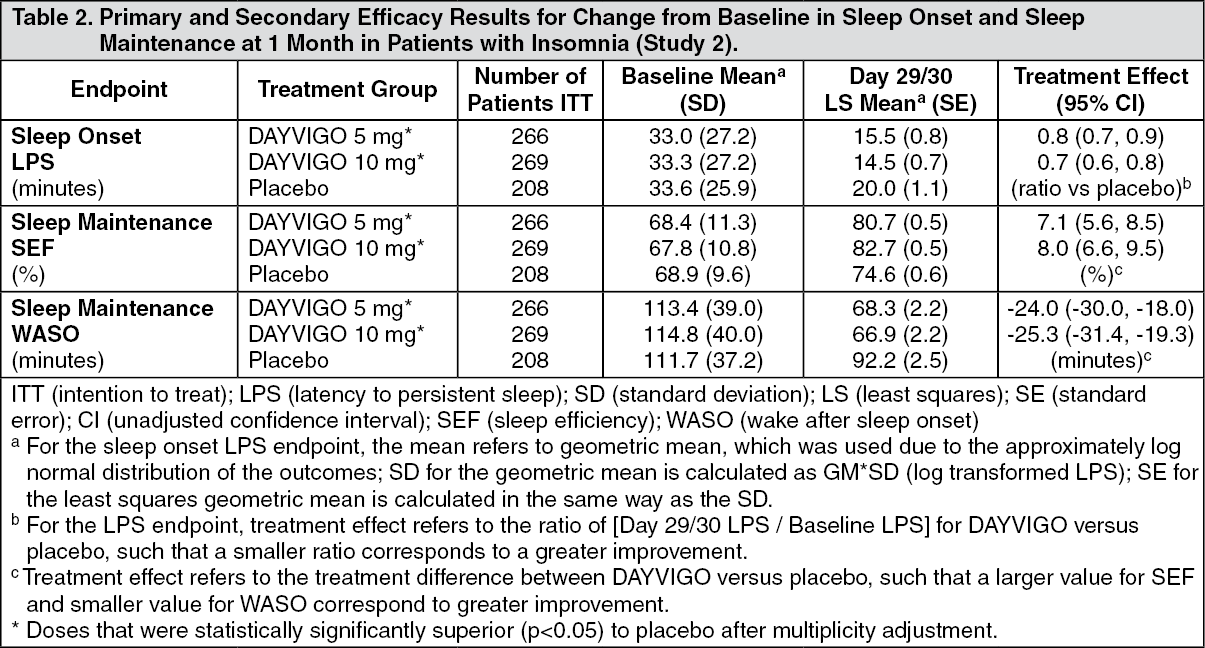 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image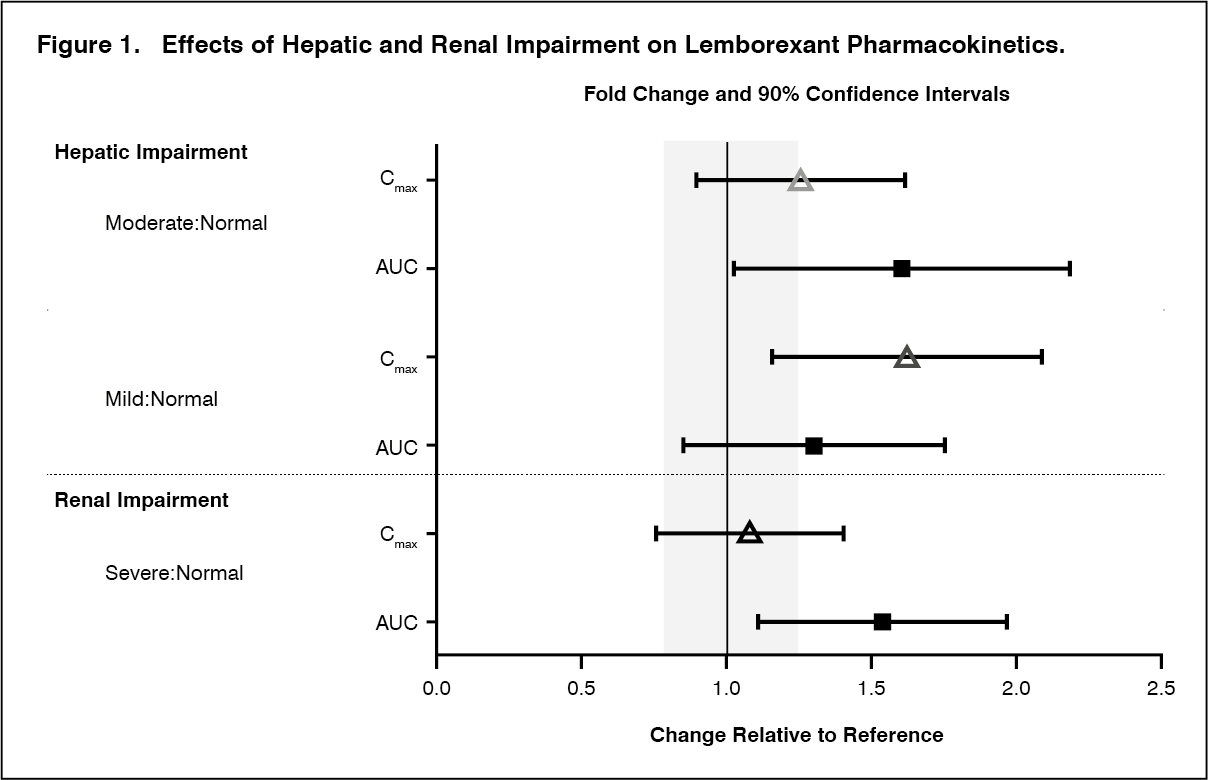 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image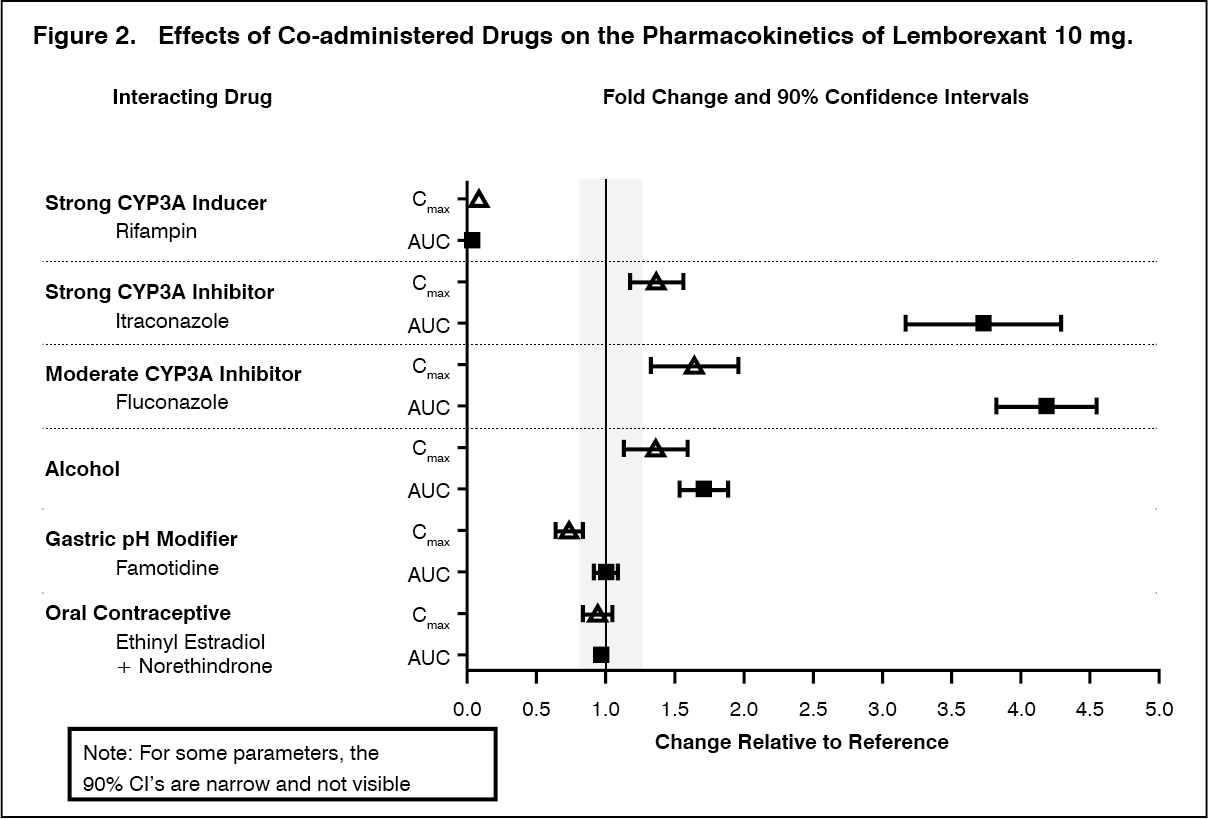 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image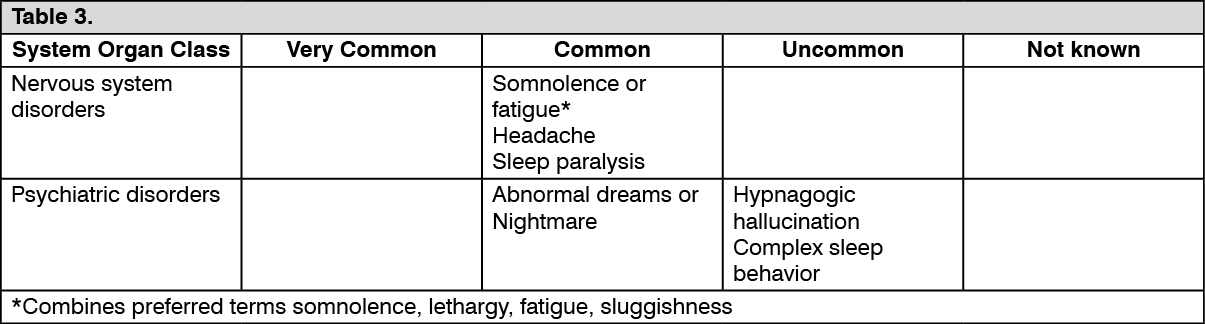 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image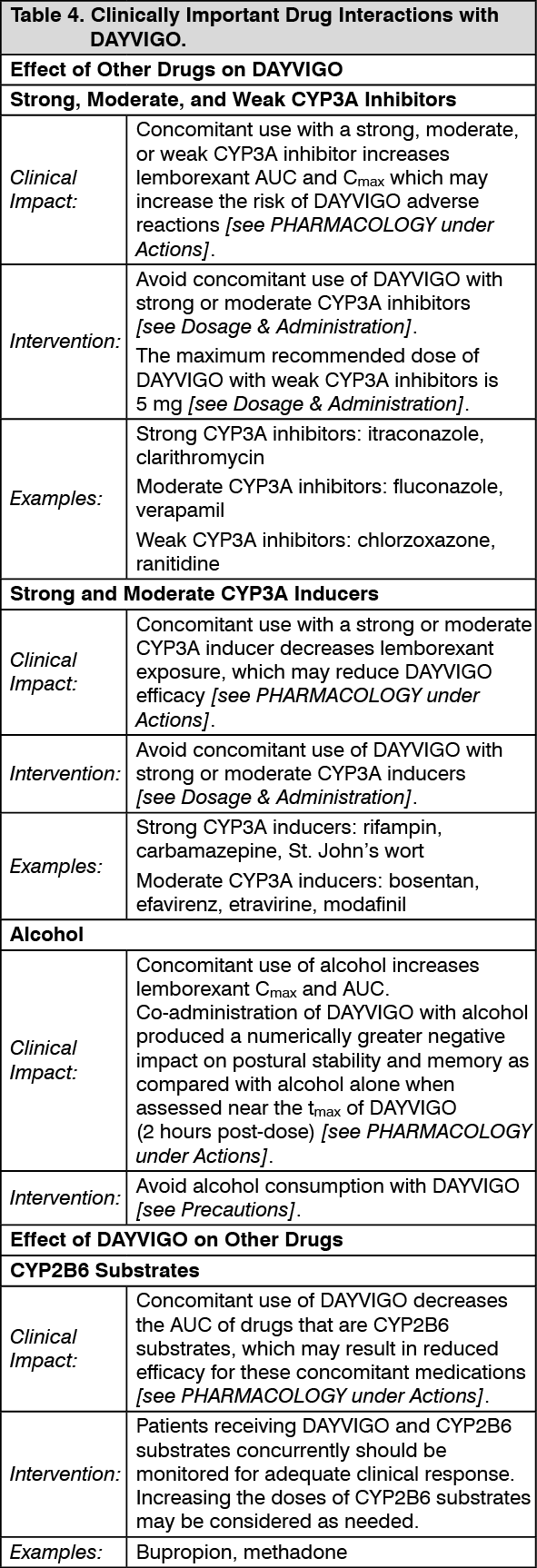 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
