Pharmacotherapeutic group: Drugs for the treatment of bone diseases, other drugs affecting bone structure and mineralisation.
ATC code: M05BX05.
Pharmacology: Pharmacodynamics: Mechanism of action: Burosumab is a recombinant human monoclonal antibody (IgG1) that binds to and inhibits the activity of fibroblast growth factor 23 (FGF23). By inhibiting FGF23, burosumab increases tubular reabsorption of phosphate from the kidney and increases serum concentration of 1,25 dihydroxy-Vitamin D.
Clinical efficacy in paediatric patients with XLH: Study UX023-CL301: In paediatric study UX023-CL301 61 patients aged 1 to 12 years (56% female; 44% male, Age at first dose, mean (SD): 6.3 (3.31) years) were randomised to burosumab (n=29) or active control (n=32; oral phosphate and active vitamin D). At entry to the study all patients had to have had a minimum of 6 months treatment of oral phosphate and active vitamin D. All patients had radiographic evidence of bone disease due to XLH (Rickets severity score ≥2). Burosumab was started at a dose of 0.8 mg/kg every 2 weeks and increased to 1.2 mg/kg if there was inadequate response, as measured by fasting serum phosphate. Those patients randomised to active control group received multiple daily doses of oral phosphate and active vitamin D.
The primary efficacy endpoint was the change in severity of rickets at Week 40, as assessed by the RGI-C (Radiographic Global Impression of change) score, compared between the burosumab and active control groups.
The RGI-C is a relative rating scale that compares a patient's rickets before and after treatment utilising a 7-point ordinal scale to evaluate change in the same abnormalities rated in the RSS (as described as follows). Scores range from -3 (indicating severe worsening of rickets) to +3 (indicating complete healing of rickets).
The severity of paediatric rickets was measured using the RSS, a radiographic scoring method based on the degree of metaphyseal fraying, concavity, and the proportion of the growth plate affected. In the UX023-CL301 study, the RSS was scored using a predefined scale looking at specific abnormalities in the wrists and knees.
All patients completed at least 64 weeks of randomised treatment, no patients had dose reductions and 8 (28%) of Burosumab-treated patients received dose escalations to 1.2 mg/kg.
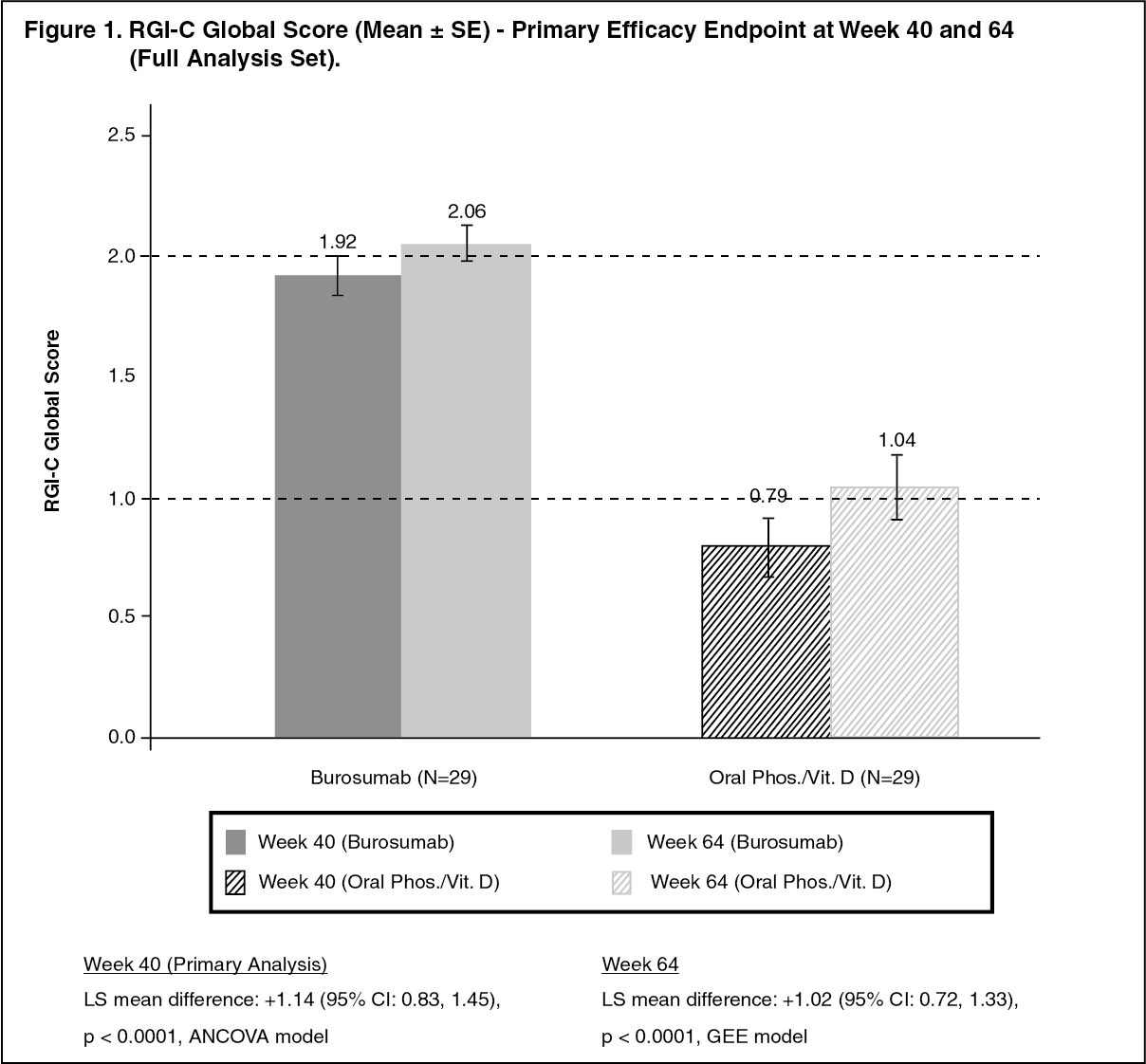
Primary Efficacy Results: Greater healing of rickets at Week 40 was seen with burosumab treatment compared to active control and this effect was maintained at week 64, as shown in Figure 1. (See Figure 1.)
 Click on icon to see table/diagram/image
Secondary Efficacy Results:
Click on icon to see table/diagram/image
Secondary Efficacy Results: Key Secondary efficacy endpoint results are presented in Table 1. (See Table 1.)
Click on icon to see table/diagram/image
Serum Phosphate: At each study visit at which serum phosphate was assessed in both groups, changes in serum phosphate from Baseline were larger in the burosumab group compared with the active control group (p < 0.0001; GEE model) (Figure 2). (See Figure 2.)
Click on icon to see table/diagram/image
Study UX023-CL201: In paediatric Study UX023-CL201, 52 paediatric patients aged 5 to 12 years (mean 8.5 years; SD 1.87) with XLH were treated for 64 weeks. Nearly all patients had radiographic evidence of rickets at baseline and had received prior oral phosphate and vitamin D analogues for a mean (SD) duration of 7 (2.4) years. This conventional therapy was discontinued 2-4 weeks prior to burosumab initiation. The burosumab dose was adjusted to target a fasting serum phosphate concentration of 3.50 to 5.02 mg/dL (1.13 to 1.62 mmol/L). Twenty six of 52 patients received burosumab every 4 weeks (Q4W). Twenty six of 52 patients received burosumab every two weeks (Q2W) at an average dose (min, max) of 0.73 (0.3, 1.5), 0.98 (0.4, 2.0) and 1.04 (0.4, 2.0) mg/kg at weeks 16, 40 and 60 respectively, and up to a maximum dose of 2.0 mg/kg.
Burosumab increased serum phosphate concentration and increased TmP/GFR. In the group that received burosumab every 2 weeks, mean (SD) serum phosphate concentration increased from 2.38 (0.405) mg/dL (0.77 (0.131) mmol/L) at baseline), to 3.3 (0.396) mg/dL (1.07 (0.128) mmol/L) at Week 40 and was maintained to Week 64 at 3.35 (0.445) mg/dL (1.08 (0.144) mmol/L).
Alkaline phosphatase activity: Mean (SD) serum total alkaline phosphatase activity was 459 (105) U/L at baseline and decreased to 369 (76) U/L at Week 64 (-19.6%, p< 0.0001).
Bone-derived serum alkaline phosphatase content was 165 (52) μg/L [mean (SD)] at Baseline and 115 (31) μg/L at Week 64 (mean change: -28.5%).
The severity of paediatric rickets in Study UX023-CL201 was measured using the RSS, as described previously. In Study UX023-CL201, the RSS was scored using a predefined scale looking at specific abnormalities in the wrists and knees. As a complement to the RSS assessment, the RGI-C rating scale was used. Results are summarised in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Study UX023-CL205: In paediatric Study UX023-CL205, burosumab was evaluated in 13 XLH patients, aged 1 to 4 years (mean 2.9 years; SD 1.1) for 40 weeks. All patients had radiographic evidence of rickets at baseline and twelve patients had received oral phosphate and vitamin D analogues for a mean (SD) duration of 16.7 (14.4) months. This conventional therapy was discontinued 2-6 weeks prior burosumab initiation. Patients received burosumab at a dose of 0.8 mg/kg every two weeks.
In Study UX023-CL205, mean (SD) fasting serum phosphate concentration increased from 2.51 (0.284) mg/dL (0.81 (0.092) mmol/L) at baseline to 3.47 (0.485) mg/dL (1.12 (0.158) mmol/L) at Week 40.
Serum alkaline phosphatase activity: Mean (SD) serum total alkaline phosphatase activity was 549 (193.8) U/L at baseline and decreased to 335 (87.6) U/L at Week 40 (mean change: -36.3%).
Rickets Severity Score (RSS): After 40 weeks of treatment with burosumab, mean total RSS improved from 2.92 (1.367) at baseline to 1.19 (0.522), corresponding to a change from baseline in LS mean (SE) change of -1.73 (0.132) (p<0.0001).
Radiographic Global Impression of Change (RGI-C): After 40 weeks of treatment with burosumab, the LS mean (SE) RGI-C Global score was +2.33 (0.08) in all 13 patients (p<0.0001) demonstrating healing of rickets. All 13 patients were considered RGI-C responders as defined by RGI-C global score ≥ +2.0.
The European Medicines Agency has deferred the obligation to submit the results of studies with burosumab in one or more subsets of the paediatric population in treatment of X-linked hypophosphataemia. See Dosage & Administration for information on paediatric use.
This medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited.
The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
Clinical efficacy in adults with XLH: Study UX023-CL303: Study UX023-CL303 is a randomised, double-blind, placebo-controlled study in 134 adult XLH patients. The study comprised of a 24-week placebo-controlled treatment phase followed by a 24-week open-label period where all patients received burosumab. Oral phosphate and active vitamin D analogues were not allowed during the study. Burosumab was administered at a dose of 1 mg/kg every 4 weeks. The primary endpoint of this study was normalisation of serum phosphate across the 24-week double-blind period. Key secondary endpoints included worst pain as measured by the Brief Pain Inventory (BPI) scale and stiffness and physical function as measured by the WOMAC (Western Ontario and McMaster Universities Osteoarthritis) Index. Exploratory endpoints included fracture and pseudofracture healing, enthesopathy, 6 Minute Walk Test, BPI Pain interference, Brief Fatigue Inventory (BFI) worst fatigue and BFI global fatigue score.
At study entry, the mean age of patients was 40 years (range 19 to 66 years) and 35% were male. 66 patients were randomised to placebo treatment and 68 to burosumab treatment; at baseline, mean (SD) serum phosphate was 0.62 (0.10) mmol/l [1.92 (0.32) mg/dL] and 0.66 (0.1 mmol/l) [2.03 (0.30) mg/dL] in the placebo and burosumab groups respectively.
For the primary efficacy endpoint, a greater proportion of patients treated with burosumab achieved a mean serum phosphate level above the lower limit of normal (LLN) compared to the placebo group through week 24 (Table 3 and Figure 3). (See Table 3 and Figure 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patient reported pain, physical function and stiffness: Change from baseline at Week 24 showed a larger difference for burosumab relative to placebo in patient reported pain (BPI), physical function (WOMAC Index) and stiffness (WOMAC Index). The mean (SE) difference between treatment groups (burosumab-placebo) reach statistical significance for WOMAC stiffness at Week 24. Details are shown in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
6 Minute Walk Test: This exercise test was conducted in all patients at Baseline, Week 12, 24, 36 and 48 (LS mean difference in change from baseline, burosumab→placebo; Table 5). Improvements continued through to Week 48 where distance walked increased from 357 m at baseline to 393 m at Week 48. Patients who crossed over from placebo to burosumab achieved similar improvements after 24 weeks of treatment. (See Table 5.)
Click on icon to see table/diagram/image
Radiographic Evaluation of Fractures and Pseudofractures: In Study UX023-CL303, a skeletal survey was conducted at baseline to identify osteomalacia-related fractures and pseudofractures. There were 52% (70/134) of patients who had either active fractures (12%, 16/134) or active pseudofractures (47%, 63/134) at baseline. Following burosumab treatment more patients showed healing of fractures and pseudofractures compared to the placebo group (Figure 4). During the placebo-controlled treatment period up to week 24, a total of 6 new fractures or pseudofractures appeared in 68 patients receiving burosumab compared to 8 new abnormalities in 66 patients receiving placebo. Of the number of new fractures developed prior to week 48 most (10/18) were healed or partially healed at the end of the study. (See Figure 4.)
Click on icon to see table/diagram/image
At Baseline, the mean (SD) total calcaneal enthesopathy burden (sum of superior and inferior calcaneal spurs) was 5.64 (3.12) cm in the burosumab group and 5.54 (3.1) cm in the placebo group. At Week 24, the mean (SD) total calcaneal enthesopathy burden was 5.90 (3.56) cm in the burosumab→burosumab group and 4.07 (2.38) cm in the placebo→burosumab group.
For the exploratory endpoints of BPI Pain interference, BFI worst fatigue and BFI global fatigue score no meaningful difference were observed between treatment arms.
Bone Histomorphometry in Adults: Study UX023-CL304: Study UX023-CL304 is a 48-week, open-label, single-arm study in adult XLH patients to assess the effects of burosumab on improvement of osteomalacia as determined by histologic and histomorphometric evaluation of iliac crest bone biopsies. Patients received 1.0 mg/kg burosumab every 4 weeks. Oral phosphate and active vitamin D analogues were not allowed during the study.
14 patients were enrolled, and at study entry, the mean age of patients was 40 years (range 25 to 52 years) and 43% were male. After 48 weeks of treatment in Study UX023-CL304 paired biopsies were available from 11 patients; healing of osteomalacia was observed in all ten evaluable patients as demonstrated by decreases in osteoid volume/bone volume (OV/BV) from a mean (SD) score of 26.1% (12.4) at baseline to 11.9% (6.6), Osteoid thickness (O.Th) declined in 11 evaluable patients from a mean (SD) of 17.2 (4.1) micrometres to 11.6 (3.1) micrometres.
Pharmacokinetics: Absorption: Burosumab absorption from subcutaneous injection sites to blood circulation is nearly complete. Following subcutaneous administration, the median time to reach maximum serum concentrations (T
max) of burosumab is approximately 7-13 days. The peak serum concentration (C
max) and area under the concentration-time curve (AUC) of serum burosumab is dose proportional over the dose range of 0.1-2.0 mg/kg.
Distribution: In XLH patients, the observed volume of distribution of burosumab approximates the volume of plasma, suggesting limited extravascular distribution.
Biotransformation: Burosumab is composed solely of amino acids and carbohydrates as a native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination: Due to its molecular size, burosumab is not expected to be directly excreted. The clearance of burosumab is dependent on body weight and estimated to be 0.290 L/day and 0.136 L/day in a typical adult (70 kg) and paediatric (30 kg) XLH patient, respectively, with corresponding disposition half-life (t½) in the serum ranging from approximately 16 to 19 days. Given the t½ estimates, the estimated time to reach the plateau of steady-state exposures is approximately 67 days. Following multiple dose administration to paediatric subjects, observed serum trough concentrations reach a plateau by 8 weeks after initiation of treatment.
Linearity/non-linearity: Burosumab displays time-invariant pharmacokinetics that is linear to dose over the subcutaneous dose range of 0.1 to 2.0 mg/kg.
Pharmacokinetic/pharmacodynamic relationship(s): With the subcutaneous route of administration, a direct PK-PD relationship between serum burosumab concentrations and increases in serum phosphate concentration is observed and well described by an E
max/EC
50 model. Serum burosumab and phosphate concentrations, as well as TmP/GFR, increased and decreased in parallel and reached maximum levels at approximately the same time point after each dose, supporting a direct PK-PD relationship. The AUC for the change from baseline in serum phosphate, TmP/GFR and 1,25(OH)
2D increased linearly with increasing burosumab AUC.
Paediatric PK/PD: No significant difference has been observed in paediatric patient pharmacokinetics or pharmacodynamics as compared with PK/PD in the adult population. Burosumab clearance and volume of distribution are body weight dependent.
Special Populations: Population PK analyses using data from paediatric and adult subjects who have XLH indicated that age, sex, race, ethnicity, baseline serum albumin, baseline serum alkaline phosphate, baseline serum alanine aminotransferase, and baseline creatinine clearance ≥49.9 mL/min, were not significant predictors of burosumab PK.
Post-Prandial Effect on Serum Phosphate and Calcium: The effect of burosumab on serum phosphate and calcium levels after food was investigated in two sub-studies (Study UX023-CL301 and UX023-CL303); 13 paediatric patients (aged >3 years) and 26 adult patients (aged 24-65 years). Serum phosphate and calcium were measured at the end of the treatment interval in paediatric patients and mid-interval in adults. Blood samples were taken after a period of fasting, and again 1-2 hours after a standardised meal.
Burosumab treatment did not cause post-prandial excursions above the age-adjusted upper limits of normal in serum phosphate or serum calcium in any paediatric or adult subject in the sub-studies.
Toxicology: Preclinical safety data: Adverse reactions in non-clinical studies with normal animals were observed at exposures which resulted in serum phosphate concentration greater than normal limits. These effects were consistent with an exaggerated response to the inhibition of normal FGF23 levels resulting in a supraphysiologic increase in serum phosphate beyond the upper limit of normal.
Studies in rabbits and adult and juvenile cynomolgus monkeys demonstrated dose-dependent elevations of serum phosphate and 1,25 (OH)
2D confirming the pharmacologic actions of burosumab in these species. Ectopic mineralisation of multiple tissues and organs (e.g. kidney, heart, lung, and aorta), and associated secondary consequences (e.g. nephrocalcinosis) in some cases, due to hyperphosphataemia, was observed in normal animals at doses of burosumab that resulted in serum phosphate concentrations in animals greater than approximately 8 mg/dL (2.6 mmol/L). In a murine model of XLH, a significant reduction in the incidence of ectopic mineralisation was observed at equivalent levels of serum phosphate, suggesting that the risk of mineralisation is less in the presence of excess FGF23.
Bone effects seen in adult and juvenile monkeys included changes in bone metabolism markers, increases in thickness and density of cortical bone, increased density of total bone and thickening of long bone. These changes were a consequence of higher than normal serum phosphate levels, which accelerated bone turnover and also led to periosteal hyperostosis and a decrease in bone strength in adult animals, but not in juvenile animals at the doses tested. Burosumab did not promote abnormal bone development, as no changes in femur length or bone strength were noted in juvenile animals. Bone changes were consistent with the pharmacology of burosumab and the role of phosphate in bone mineralization, metabolism and turnover.
In repeat-dose toxicology studies of up to 40 weeks duration in cynomolgus monkeys, mineralisation of the rete testis/seminiferous tubules was observed in male monkeys; however, no changes were observed in semen analysis. No adverse effects on female reproductive organs were observed in these studies.
In the reproductive and developmental toxicology study performed in pregnant cynomolgus monkeys, moderate mineralisation of the placenta was seen in pregnant animals given 30 mg/kg of burosumab and occurred in animals with peak serum phosphate concentration greater than approximately 8 mg/dL (2.6 mmol/L). Shortening of the gestation period and associated increased incidence of premature births were observed in pregnant monkeys at doses of ≥ 0.3 mg/kg which corresponded to burosumab exposures that are ≥0.875- to 1.39-fold anticipated clinical levels. Burosumab was detected in serum from fetuses indicating that burosumab was transported across the placenta to the fetus. There was no evidence of teratogenic effects. Ectopic mineralisation was not observed in foetuses or offspring and burosumab did not affect pre- and postnatal growth including survivability of the offspring.
In preclinical studies, ectopic mineralisation has been observed in normal animals, most frequently in the kidney, given burosumab at doses that resulted in serum phosphate concentrations greater than 8 mg/dL (2.6 mmol/L). Neither new or clinically meaningful worsening of nephrocalcinosis nor ectopic mineralisation have been observed in clinical trials of patients with XLH treated with Burosumab to achieve normal serum phosphate levels.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image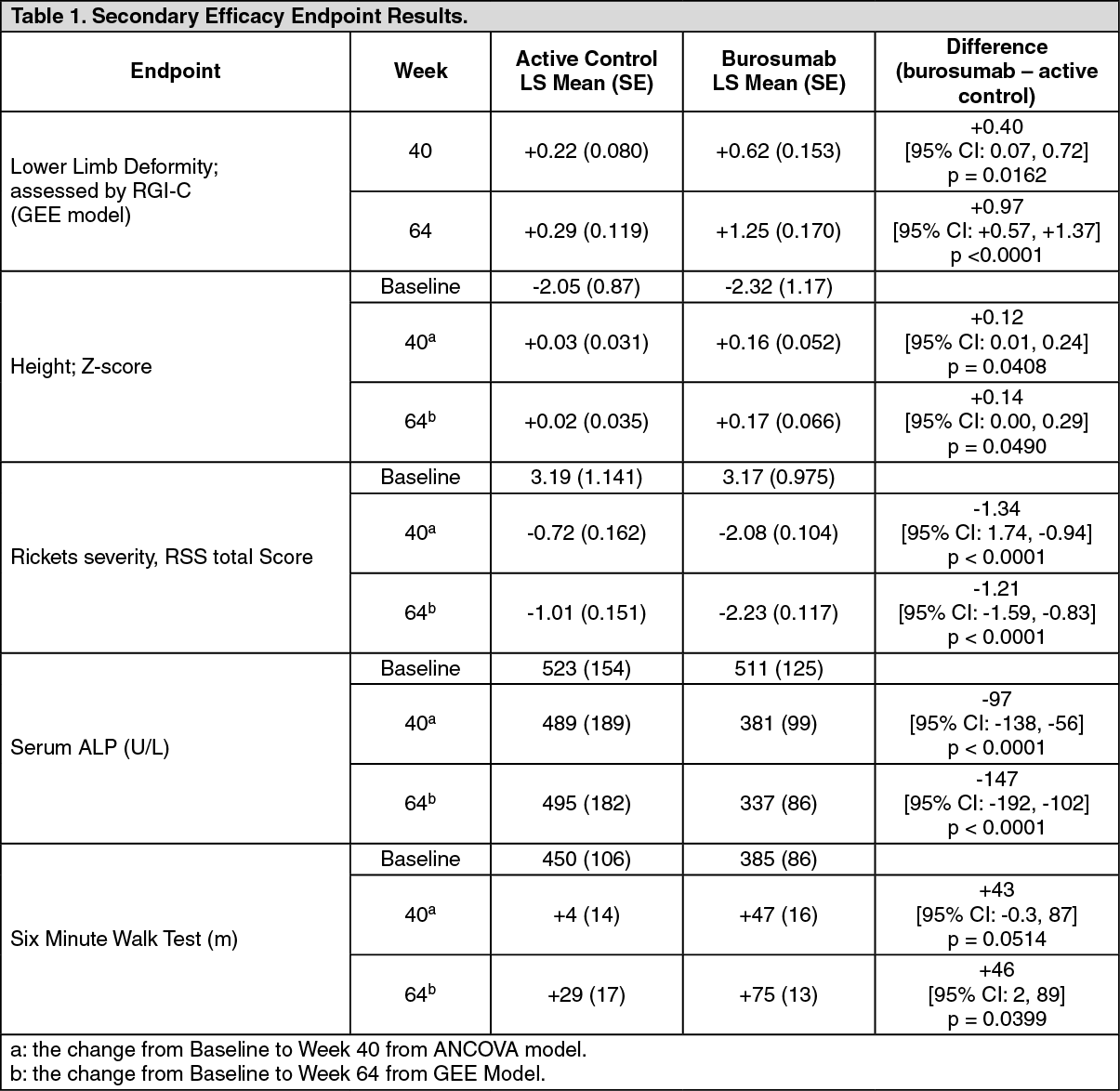 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image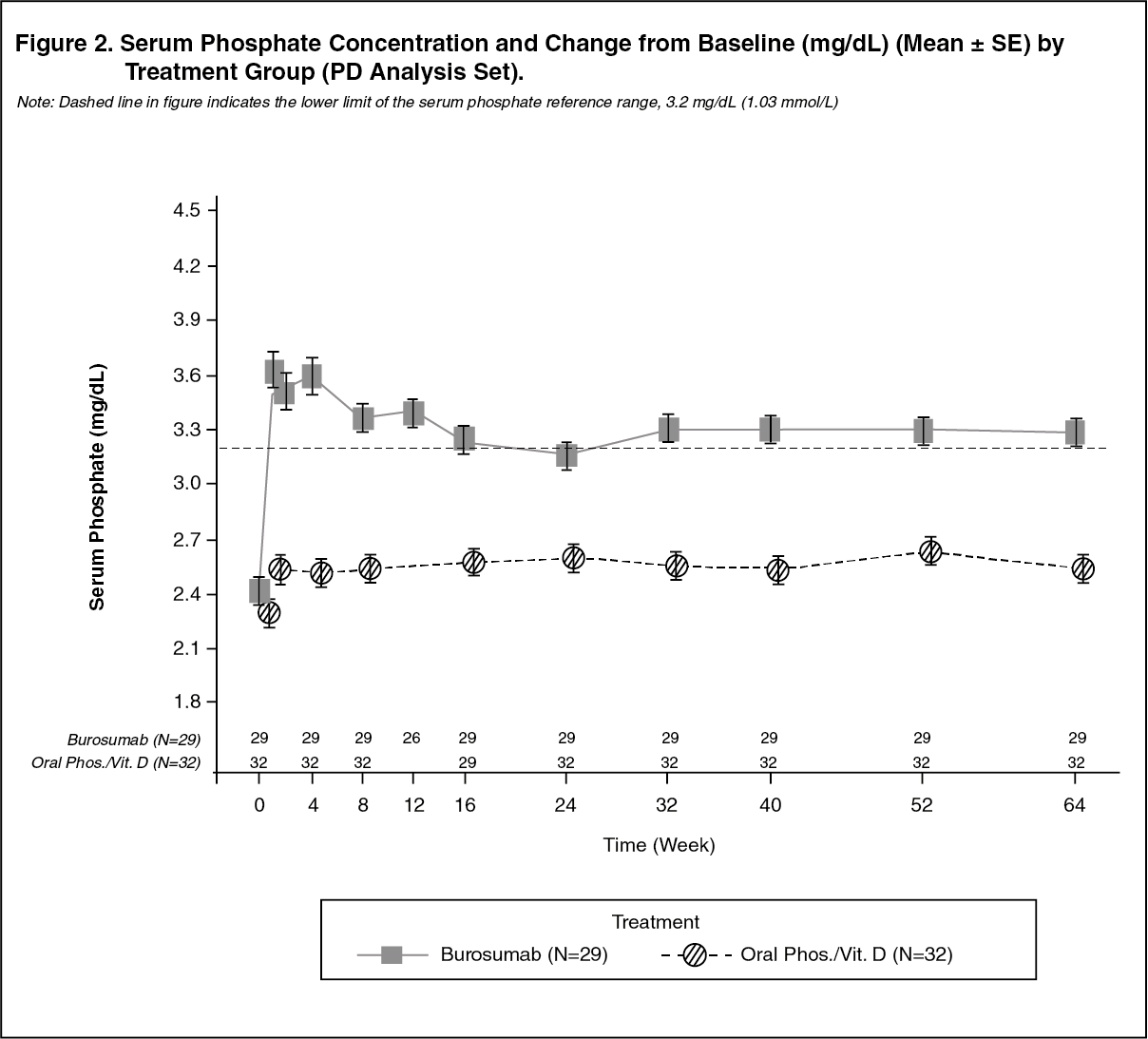 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image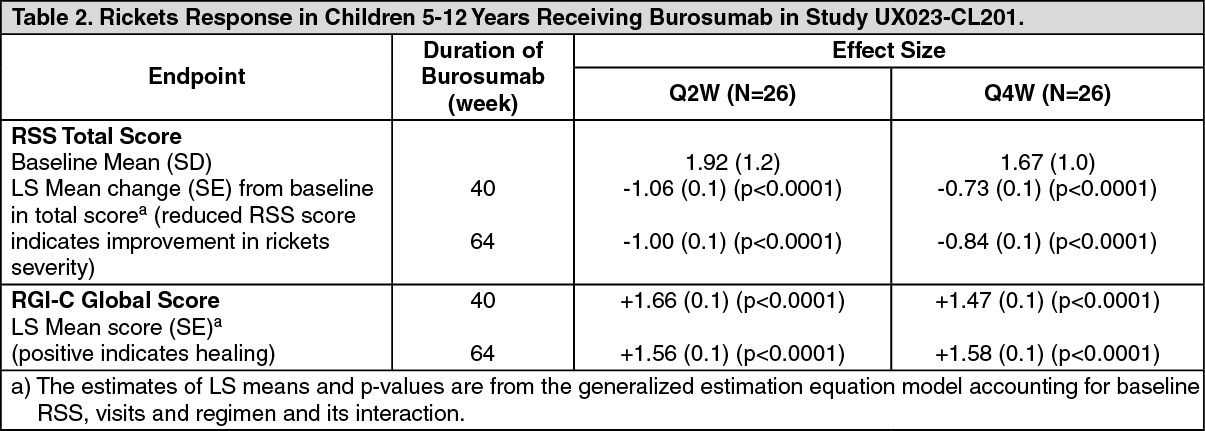 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image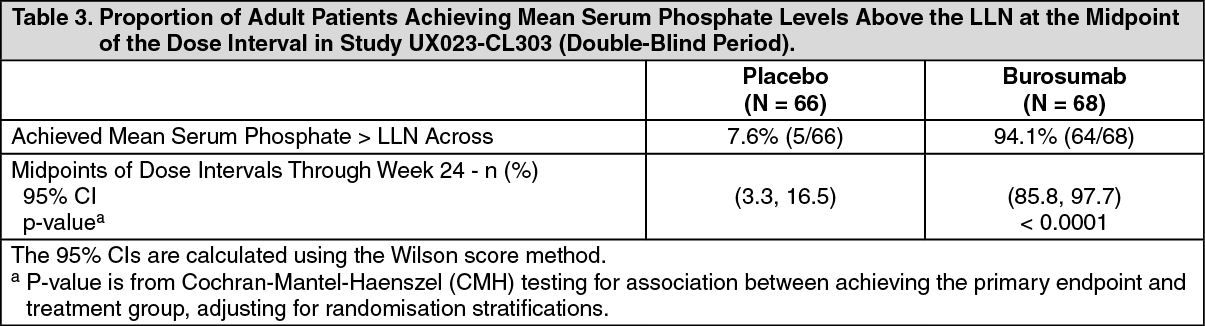 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image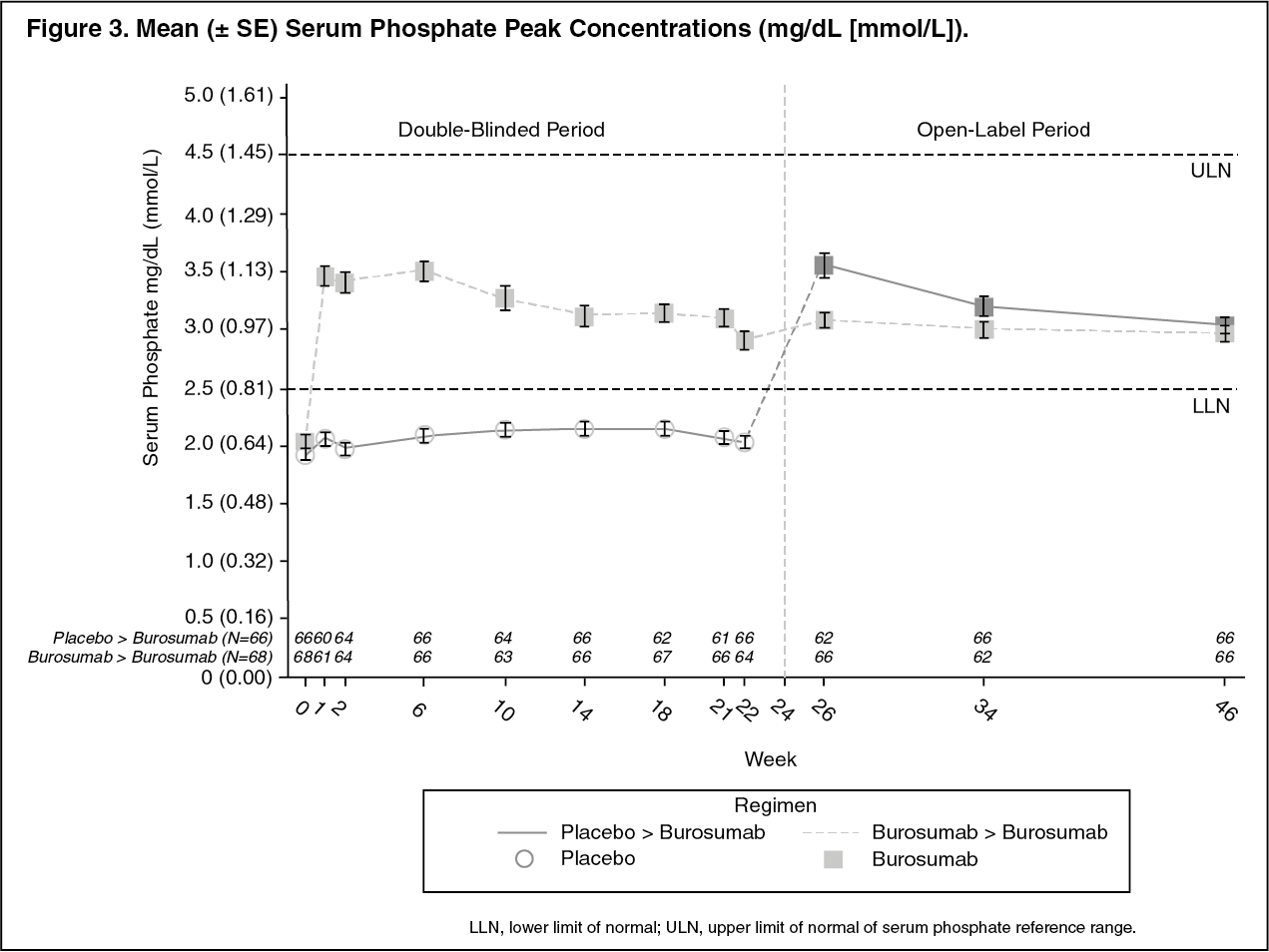 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image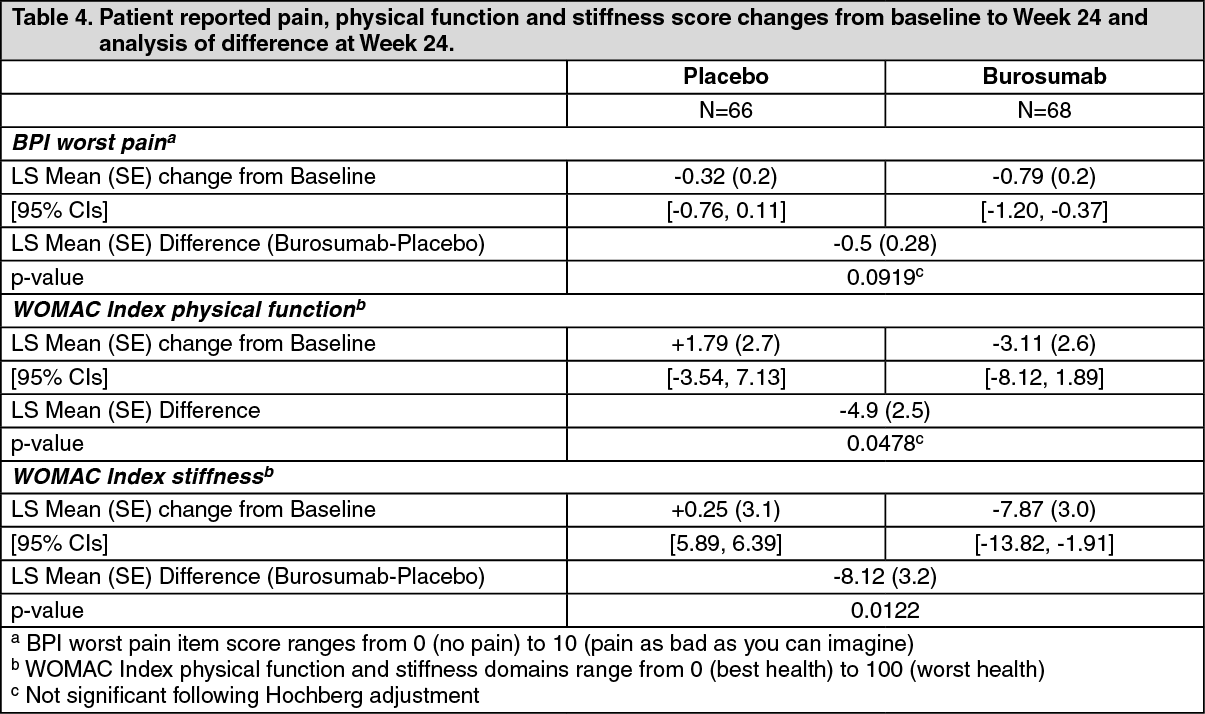 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image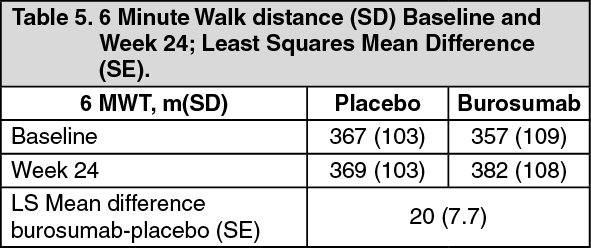 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image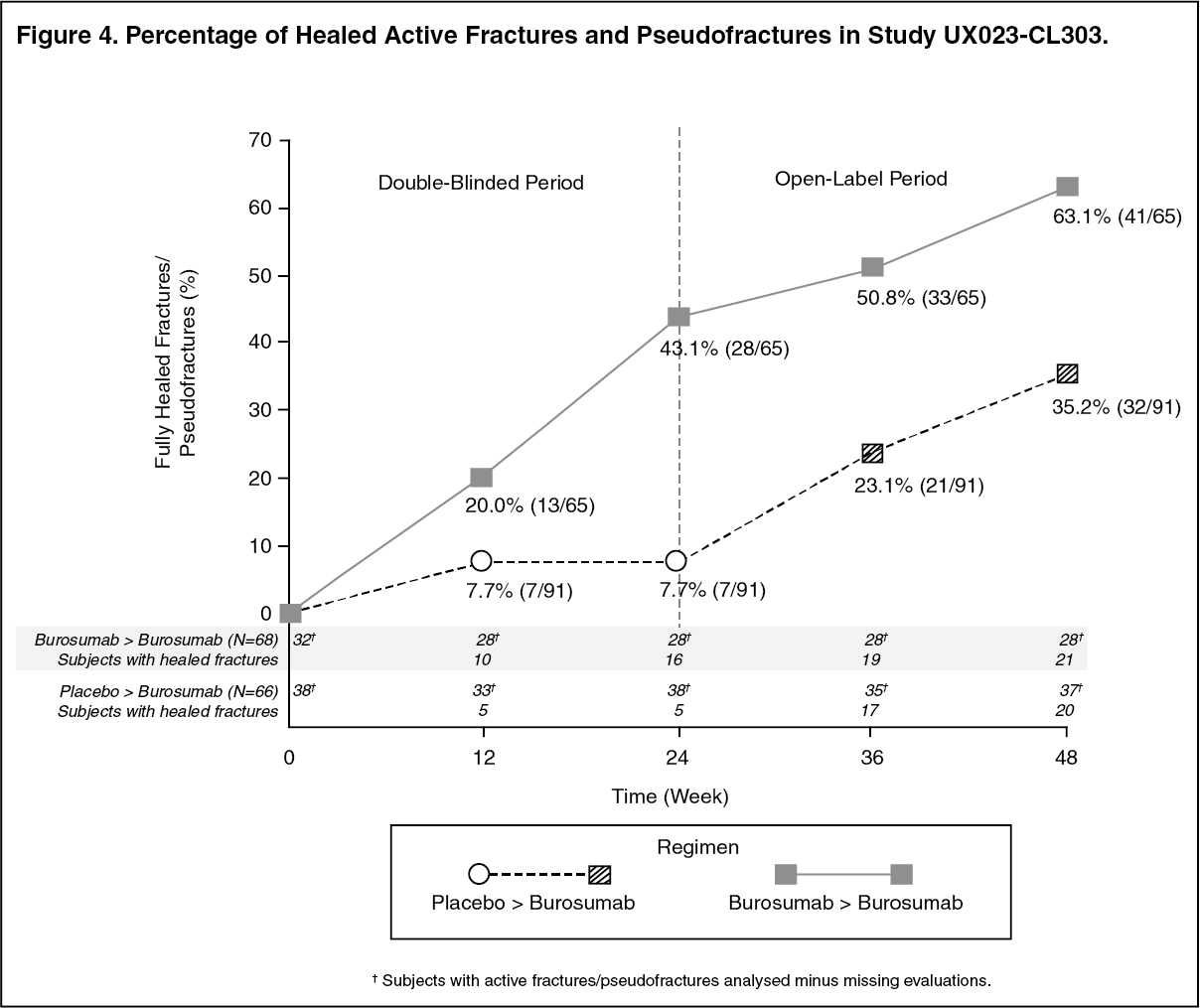 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image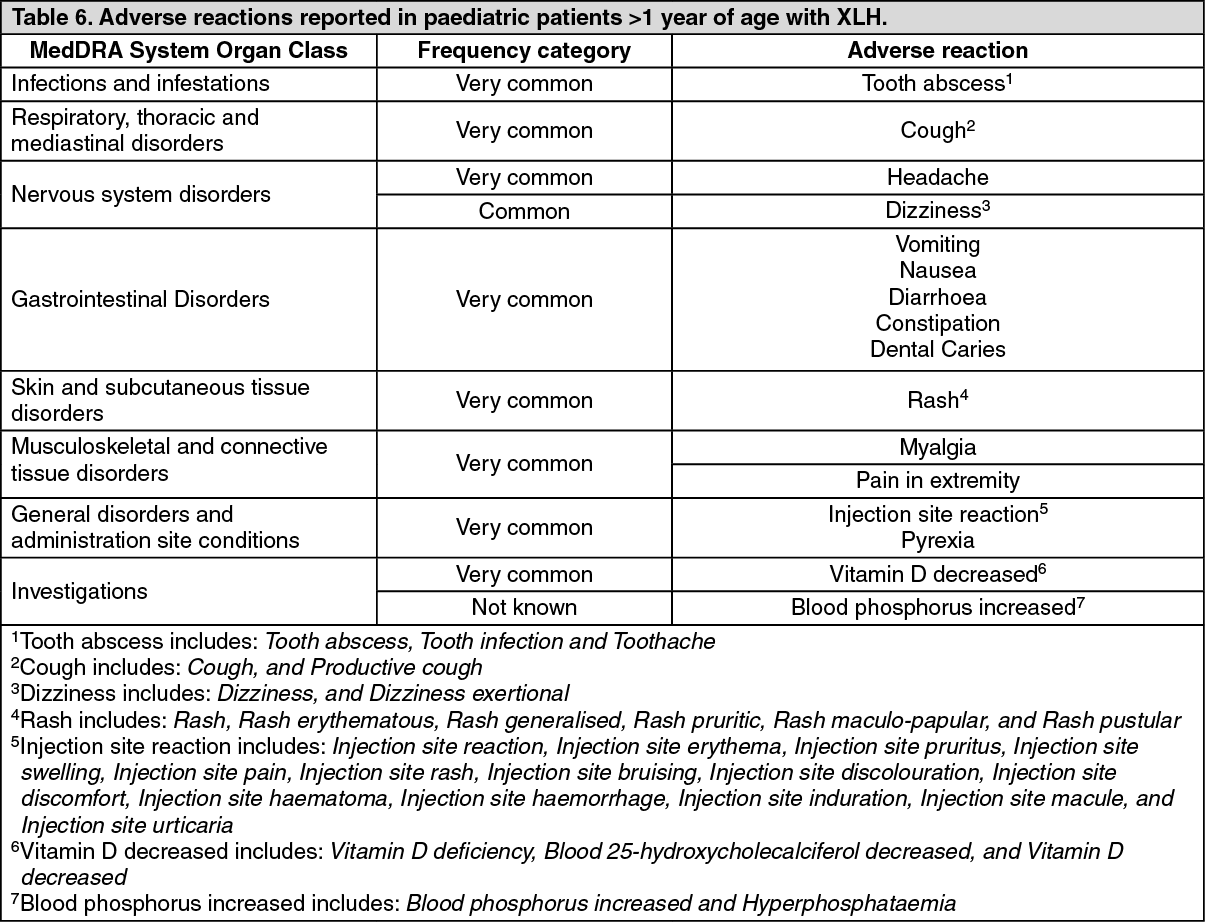 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image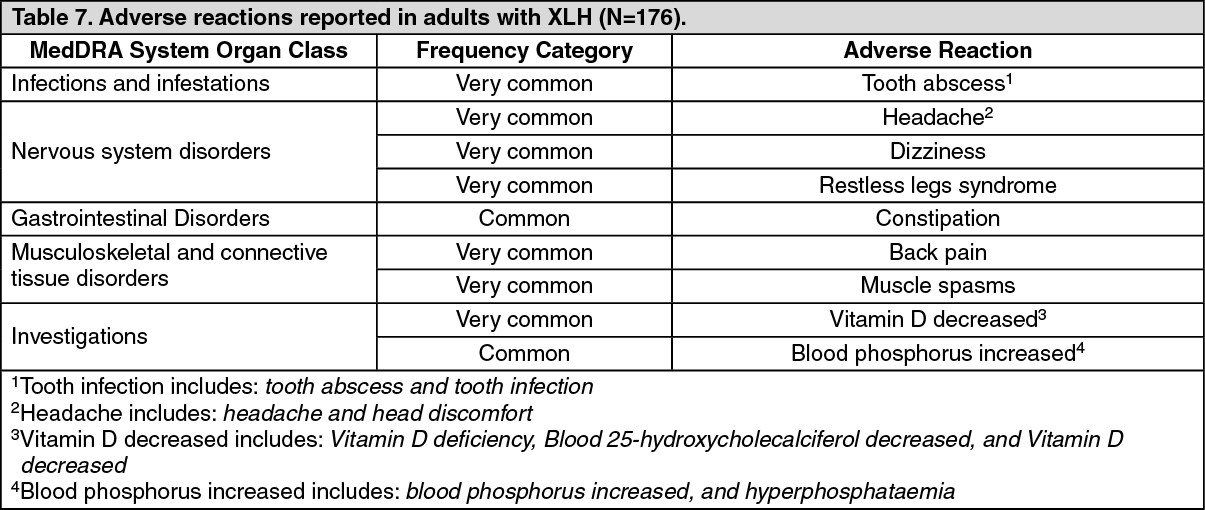 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
