Pharmacotherapeutic group: Antineoplastic agents, pyrimidine analogues.
ATC code: L01BC07.
Pharmacology: Pharmacodynamics: Mechanism of action: Azacitidine is believed to exert its antineoplastic effects by multiple mechanisms including cytotoxicity on abnormal haematopoietic cells in the bone marrow and hypomethylation of DNA. The cytotoxic effects of azacitidine may result from multiple mechanisms, including inhibition of DNA, RNA and protein synthesis, incorporation into RNA and DNA, and activation of DNA damage pathways. Non-proliferating cells are relatively insensitive to azacitidine. Incorporation of azacitidine into DNA results in the inactivation of DNA methyltransferases, leading to hypomethylation of DNA. DNA hypomethylation of aberrantly methylated genes involved in normal cell cycle regulation, differentiation and death pathways may result in gene re-expression and restoration of cancer-suppressing functions to cancer cells. The relative importance of DNA hypomethylation versus cytotoxicity or other activities of azacitidine to clinical outcomes has not been established.
Clinical efficacy and safety: Adult population (MDS, CMML and AML [20-30% marrow blasts]): The efficacy and safety of Azacitidine for Injection 100 mg/vial were studied in an international, multicenter, controlled, open-label, randomised, parallel-group, Phase 3 comparative study (AZA PH GL 2003 CL 001) in adult patients with: intermediate-2 and high-risk MDS according to the International Prognostic Scoring System (IPSS), refractory anaemia with excess blasts (RAEB), refractory anaemia with excess blasts in transformation (RAEB-T) and modified chronic myelomonocytic leukaemia (mCMML) according to the French American British (FAB) classification system. RAEB-T patients (21-30% blasts) are now considered to be AML patients under the current WHO classification system. Azacitidine plus best supportive care (BSC) (n=179) was compared to conventional care regimens (CCR). CCR consisted of BSC alone (n=105), low-dose cytarabine plus BSC (n=49) or standard induction chemotherapy plus BSC (n=25). Patients were pre-selected by their physician to 1 of the 3 CCR prior to randomisation. Patients received this pre-selected regimen if not randomised to Azacitidine for Injection 100 mg/vial. As part of the inclusion criteria, patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2. Patients with secondary MDS were excluded from the study. The primary endpoint of the study was overall survival. Azacitidine for Injection 100 mg/vial was administered at a subcutaneous dose of 75 mg/m
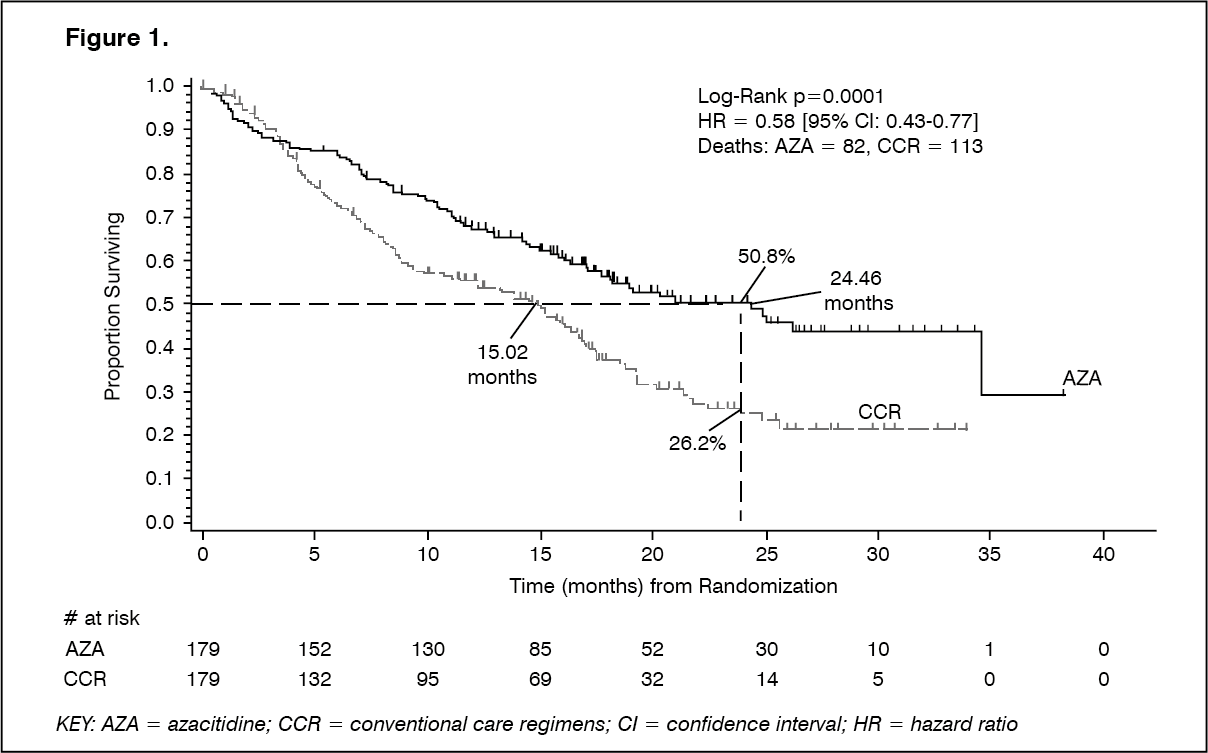
2 daily for 7 days, followed by a rest period of 21 days (28-day treatment cycle) for a median of 9 cycles (range=1-39) and a mean of 10.2 cycles. Within the Intent to Treat population (ITT), the median age was 69 years (range 38 to 88 years). In the ITT analysis of 358 patients (179 azacitidine and 179 CCR), Azacitidine for Injection 100 mg/vial treatment was associated with a median survival of 24.46 months versus 15.02 months for those receiving CCR treatment, a difference of 9.4 months, with a stratified log-rank p-value of 0.0001. The hazard ratio for the treatment effect was 0.58 (95% CI: 0.43, 0.77). The two year survival rates were 50.8% in patients receiving azacitidine versus 26.2% in patients receiving CCR (p<0.0001). (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The survival benefits of Azacitidine for Injection 100 mg/vial were consistent regardless of the CCR treatment option BSC alone, low-dose cytarabine plus BSC or standard induction chemotherapy plus BSC) utilised in the control arm.
When IPSS cytogenetic subgroups were analysed, similar findings in terms of median overall survival were observed in all groups (good, intermediate, poor cytogenetics, including monosomy 7).
On analyses of age subgroups, an increase in median overall survival was observed for all groups (<65 years, ≥65 years and ≥75 years).
Azacitidine for Injection 100 mg/vial treatment was associated with a median time to death or transformation to AML of 13.0 months versus 7.6 months for those receiving CCR treatment, an improvement of 5.4 months with a stratified log-rank p-value of 0.0025.
Azacitidine for Injection 100 mg/vial treatment was also associated with a reduction in cytopenias, and their related symptoms. Azacitidine for Injection 100 mg/vial treatment led to a reduced need for red blood cell (RBC) and platelet transfusions. Of the patients in the azacitidine group who were RBC transfusion dependent at baseline, 45.0% of these patients became RBC transfusion independent during the treatment period, compared with 11.4% of the patients in the combined CCR groups (a statistically significant p<0.0001) difference of 33.6% (95% CI: 22.4, 44.6). In patients who were RBC transfusion dependent at baseline and became independent, the median duration of RBC transfusion independence was 13 months in the azacitidine group.
Response was assessed by the investigator or by the Independent Review Committee (IRC). Overall response (complete remission [CR] + partial remission [PR]) as determined by the investigator was 29% in the azacitidine group and 12% in the combined CCR group (p=0.0001). Overall response (CR + PR) as determined by the IRC in AZA PH GL 2003 CL 001 was 7% (12/179) in the azacitidine group compared with 1% (2/179) in the combined CCR group (p=0.0113). The differences between the IRC and investigator assessments of response were a consequence of the International Working Group (IWG) criteria requiring improvement in peripheral blood counts and maintenance of these improvements for a minimum of 56 days. A survival benefit was also demonstrated in patients that had not achieved a complete/partial response following azacitidine treatment. Haematological improvement (major or minor) as determined by the IRC was achieved in 49% of patients receiving azacitidine compared with 29% of patients treated with combined CCR (p <0.0001).
In patients with one or more cytogenetic abnormalities at baseline, the percentage of patients with a major cytogenetic response was similar in the azacitidine and combined CCR groups. Minor cytogenetic response was statistically significantly (p=0.0015) higher in the azacitidine group (34%) compared with the combined CCR group (10%).
Adult population aged 65 years or older with AML with >30% marrow blasts: The results presented as follows represent the intent-to-treat population studied in AZA-AML-001 (see Indications/Uses for the approved indication).
The efficacy and safety of Azacitidine for Injection 100 mg/vial was studied in an international, multicentre, controlled, open-label, parallel group Phase 3 study in patients 65 years and older with newly diagnosed de novo or secondary AML with >30% bone marrow blasts according to the WHO classification, who were not eligible for HSCT. Azacitidine for Injection 100 mg/vial plus BSC (n=241) was compared to CCR. CCR consisted of BSC alone (n=45), low-dose cytarabine plus BSC (n=158), or standard intensive chemotherapy with cytarabine and anthracycline plus BSC (n=44). Patients were pre-selected by their physician to 1 of the 3 CCRs prior to randomization. Patients received the preselected regimen if not randomised to Azacitidine for Injection 100 mg/vial. As part of the inclusion criteria, patients were required to have an ECOG performance status of 0-2 and intermediate- or poor-risk cytogenetic abnormalities. The primary endpoint of the study was overall survival.
Azacitidine for Injection 100 mg/vial was administered at a SC dose of 75 mg/m
2/day for 7 days, followed by a rest period of 21 days (28 day treatment cycle), for a median of 6 cycles (range: 1 to 28), BSC-only patients for a median of 3 cycles (range: 1 to 20), low-dose cytarabine patients for a median of 4 cycles (range 1 to 25) and standard intensive chemotherapy patients for a median of 2 cycles (range: 1 to 3, induction cycle plus 1 or 2 consolidation cycles).
The individual baseline parameters were comparable between the Azacitidine for Injection 100 mg/vial and CCR groups. The median age of the subjects was 75.0 years (range: 64 to 91 years), 75.2% were Caucasian and 59.0% were male. At baseline 60.7% were classified as AML not otherwise specified, 32.4% AML with myelodysplasia-related changes, 4.1% therapy-related myeloid neoplasms and 2.9% AML with recurrent genetic abnormalities according to the WHO classification.
In the ITT analysis of 488 patients (241 Azacitidine for Injection 100 mg/vial and 247 CCR), Azacitidine for Injection 100 mg/vial treatment was associated with a median survival of 10.4 months versus 6.5 months for those receiving CCR treatment, a difference of 3.8 months, with a stratified log-rank p-value of 0.1009 (two-sided). The hazard ratio for the treatment effect was 0.85 (95% CI=0.69, 1.03). The one-year survival rates were 46.5% in patients receiving Azacitidine for Injection 100 mg/vial versus 34.3% in patients receiving CCR. (See Figure 2.)
Click on icon to see table/diagram/image
The Cox PH model adjusted for pre-specified baseline prognostic factors defined a HR for Azacitidine for Injection 100 mg/vial versus CCR of 0.80 (95% CI=0.66, 0.99; p=0.0355).
In addition, although the study was not powered to demonstrate a statistically significant difference when comparing azacitidine to the preselection CCR treatment groups, the survival of Azacitidine for Injection 100 mg/vial treated patients was longer when compared to CCR treatment options BSC alone, low-dose cytarabine plus BSC and were similar when compared to standard intensive chemotherapy plus BSC.
In all pre-specified subgroups age [(<75 years & ≥75 years), gender, race, ECOG performance status (0 or 1 & 2), baseline cytogenetic risk (intermediate & poor), geographic region, WHO classification of AML (including AML with myelodysplasia-related changes), baseline WBC count (≤5 x 10
9/L & >5 x 10
9/L), baseline bone marrow blasts (≤50% & >50%) and prior history of MDS] there was a trend in OS benefit in favour of Azacitidine for Injection 100 mg/vial. In a few pre-specified subgroups, the OS HR reached statistical significance including patients with poor cytogenetic risk, patients with AML with myelodysplasia-related changes, patients <75 years, female patients and white patients.
Haematologic and cytogenetic responses were assessed by the investigator and by the IRC with similar results. Overall response rate (complete remission [CR] + complete remission with incomplete blood count recovery [CRi]) as determined by the IRC was 27.8% in the Azacitidine for Injection 100 mg/vial group and 25.1% in the combined CCR group (p=0.5384). In patients who achieved CR or CRi, the median duration of remission was 10.4 months (95% CI=7.2, 15.2) for the Azacitidine for Injection 100 mg/vial subjects and 12.3 months (95% CI=9.0, 17.0) for the CCR subjects. A survival benefit was also demonstrated in patients that had not achieved a complete response for Azacitidine for Injection 100 mg/vial compared to CCR.
Azacitidine for Injection 100 mg/vial treatment improved peripheral blood counts and led to a reduced need for RBC and platelet transfusions. A patient was considered RBC or platelet transfusion dependent at baseline if the subject had one or more RBC or platelet transfusions during the 56 days (8 weeks) on or prior to randomization, respectively. A patient was considered RBC or platelet transfusion independent during the treatment period if the subject had no RBC or platelet transfusions during any consecutive 56 days during the reporting period, respectively.
Of the patients in the Azacitidine for Injection 100 mg/vial group who were RBC transfusion dependent at baseline, 38.5% (95% CI=31.1, 46.2) of these patients became RBC transfusion independent during the treatment period, compared with 27.6% of (95% CI=20.9, 35.1) patients in the combined CCR groups. In patients who were RBC transfusion dependent at baseline and achieved transfusion independence on treatment, the median duration of RBC transfusion independence was 13.9 months in the Azacitidine for Injection 100 mg/vial group and was not reached in the CCR group.
Of the patients in the Azacitidine for Injection 100 mg/vial group who were platelet transfusion dependent at baseline, 40.6% (95% CI=30.9, 50.8) of these patients became platelet transfusion independent during the treatment period, compared with 29.3% of (95% CI=19.7, 40.4) patients in the combined CCR groups. In patients who were platelet transfusion dependent at baseline and achieved transfusion independence on treatment, the median duration of platelet transfusion independence was 10.8 months in the Azacitidine for Injection 100 mg/vial group and 19.2 months in the CCR group. Health-Related Quality of Life (HRQoL) was assessed using the European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire (EORTC QLQ-C30). HRQoL data could be analysed for a subset of the full trial population. While there are limitations in the analysis, the available data suggest that patients do not experience meaningful deterioration in quality of life during treatment with Azacitidine for Injection 100 mg/vial.
Pharmacokinetics: Absorption: Following subcutaneous administration of a single 75 mg/m
2 dose, azacitidine was rapidly absorbed with peak plasma concentrations of 750 ± 403 ng/mL occurring at 0.5 h after dosing (the first sampling point). The absolute bioavailability of azacitidine after subcutaneous relative to intravenous administration (single 75 mg/m
2 doses) was approximately 89% based on area under the curve (AUC).
Area under the curve and maximum plasma concentration (C
max) of subcutaneous administration of azacitidine were approximately proportional within the 25 to 100 mg/m
2 dose range.
Distribution: Following intravenous administration, the mean volume of distribution was 76 ± 26 L, and systemic clearance was 147 ± 47 L/h.
Biotransformation: Based on in vitro data, azacitidine metabolism does not appear to be mediated by cytochrome P450 isoenzymes (CYPs), UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), and glutathione transferases (GSTs).
Azacitidine undergoes spontaneous hydrolysis and deamination mediated by cytidine deaminase. In human liver S9 fractions, formation of metabolites was independent of NADPH implying that azacitidine metabolism was not mediated by cytochrome P450 isoenzymes. An in vitro study of azacitidine with cultured human hepatocytes indicates that at concentrations of 1.0 μM to 100 μM (i.e. up to approximately 30-fold higher than clinically achievable concentrations), azacitidine does not induce CYP 1A2, 2C19, or 3A4 or 3A5. In studies to assess inhibition of a series of P450 isoenzymes (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4) azacitidine up to 100 μM did not produce inhibition. Therefore, CYP enzyme induction or inhibition by azacitidine at clinically achievable plasma concentrations is unlikely.
Elimination: Azacitidine is cleared rapidly from plasma with a mean elimination half-life (t½) after subcutaneous administration of 41 ± 8 minutes. No accumulation occurs after subcutaneous administration of 75 mg/m
2 azacitidine once daily for 7 days. Urinary excretion is the primary route of elimination of azacitidine and/or its metabolites. Following intravenous and subcutaneous administration of 14C-azacitidine, 85 and 50% of the administered radioactivity was recovered in urine respectively, while <1% was recovered in faeces.
Special populations: The effects of hepatic impairment (see Dosage & Administration), gender, age, or race on the pharmacokinetics of azacitidine have not been formally studied.
Renal impairment: Renal impairment has no major effect on the pharmacokinetic exposure of azacitidine after single and multiple subcutaneous administrations. Following subcutaneous administration of a single 75 mg/m
2 dose, mean exposure values (AUC and C
max) from subjects with mild, moderate and severe renal impairment were increased by 11-21%, 15-27%, and 41-66%, respectively, compared to normal renal function subjects. However, exposure was within the same general range of exposures observed for subjects with normal renal function. Azacitidine can be administered to patients with renal impairment without initial dose adjustment provided these patients are monitored for toxicity since azacitidine and/or its metabolites are primarily excreted by the kidney.
Pharmacogenomics: The effect of known cytidine deaminase polymorphisms on azacitidine metabolism has not been formally investigated.
Toxicology: Preclinical safety data: Azacitidine induces both gene mutations and chromosomal aberrations in bacterial and mammalian cell systems
in vitro. The potential carcinogenicity of azacitidine was evaluated in mice and rats. Azacitidine induced tumours of the haematopoietic system in female mice, when administered intraperitoneally 3 times per week for 52 weeks. An increased incidence of tumours in the lymphoreticular system, lung, mammary gland, and skin was seen in mice treated with azacitidine administered intraperitoneally for 50 weeks. A tumorigenicity study in rats revealed an increased incidence of testicular tumours.
Early embryotoxicity studies in mice revealed a 44% frequency of intrauterine embryonal death (increased resorption) after a single intraperitoneal injection of azacitidine during organogenesis. Developmental abnormalities in the brain have been detected in mice given azacitidine on or before closure of the hard palate. In rats, azacitidine caused no adverse reactions when given pre-implantation, but it was clearly embryotoxic when given during organogenesis. Foetal abnormalities during organogenesis in rats included: CNS anomalies (exencephaly/encephalocele), limb anomalies (micromelia, club foot, syndactyly, oligodactyly) and others (microphthalmia, micrognathia, gastroschisis, oedema, and rib abnormalities).
Administration of azacitidine to male mice prior to mating with untreated female mice resulted in decreased fertility and loss of offspring during subsequent embryonic and postnatal development. Treatment of male rats resulted in decreased weight of the testes and epididymides, decreased sperm counts, decreased pregnancy rates, an increase in abnormal embryos and increased loss of embryos in mated females (see Precautions).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image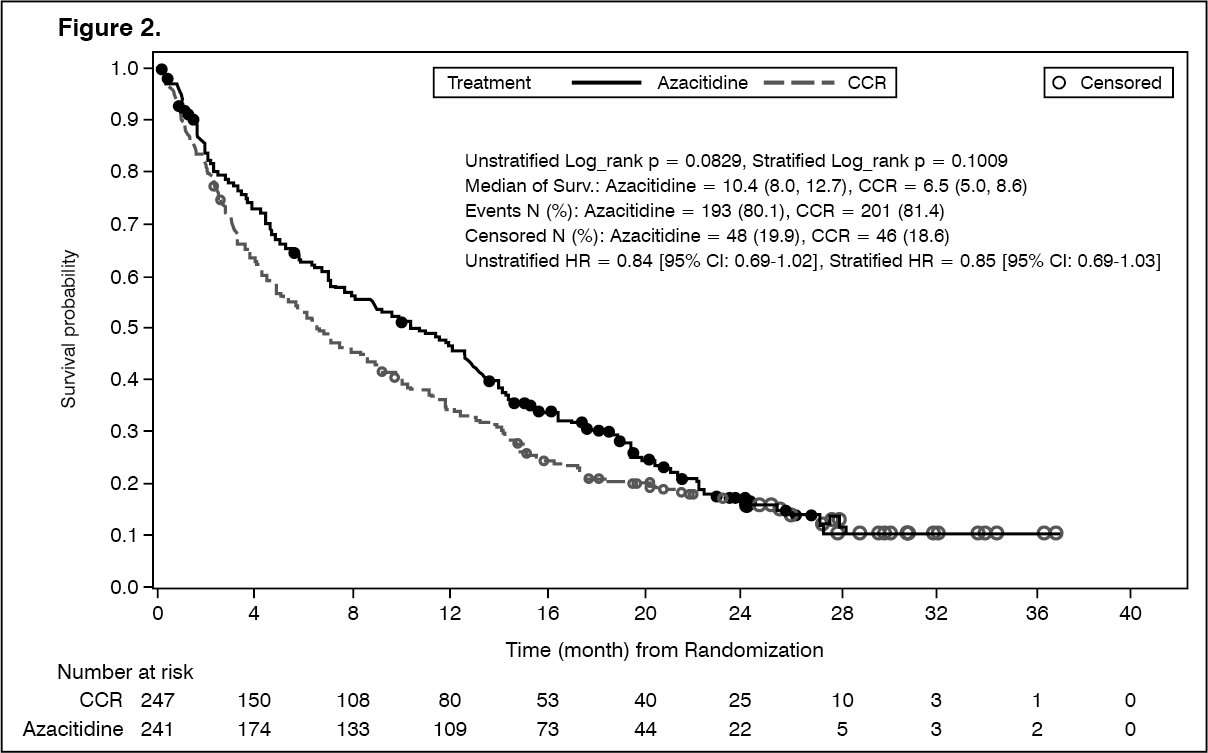 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image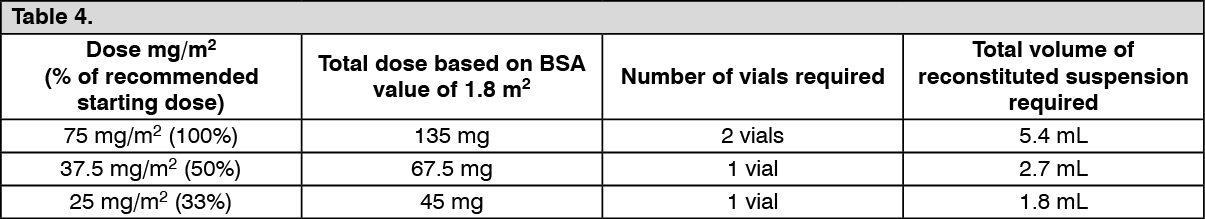 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
