Monitor carefully patients for signs and symptoms of hypersensitivity reactions during and following each administration of Venofer.
Venofer should only be administered when staff trained to evaluate and manage anaphylactic reactions is immediately available, in an environment where full resuscitation facilities can be assured. The patient should be observed for adverse effects for at least 30 minutes following each Venofer injection.
Posology:
The cumulative dose of Venofer must be calculated for each patient individually and must not be exceeded.
Calculation of dosage: The total cumulative dose of Venofer, equivalent to the total iron deficit (mg), is determined by the haemoglobin level (Hb) and body weight (BW). The dose of Venofer must be individually calculated for each patient according to the total iron deficit calculated with the following Ganzoni formula, for example: (See Equation 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Total amount of Venofer to be administered according to body weight, actual Hb level and target Hb level
*: (See Table 1.)
Click on icon to see table/diagram/image
To convert Hb (mM) to Hb (g/dl), multiply the former by 1.6.
If the total necessary dose exceeds the maximum allowed single dose, then the administration must be divided. If no response of the haematological parameters is observed after 1 to 2 weeks the original diagnosis should be reconsidered.
Calculation of dosage for iron replacement secondary to blood loss and to support autologous blood donation: The required Venofer dose to compensate for the iron deficit may be calculated according to the following formulas: If the quantity of blood lost is known: The administration of 200 mg iron (10 ml of Venofer) should result in an increase in Hb approximately equivalent to 1 unit blood (400 ml with Hb = 15 g/dl). (See Equation 2.)
Click on icon to see table/diagram/image
If the Hb level is less than desired: Formula assumes that the storage iron does not need to be restored. (See Equation 3.)
Click on icon to see table/diagram/image
For the maximum tolerated single and weekly dose, see "Normal posology" and "Maximum tolerated single and weekly doses" as follows.
Normal posology: Adults: 5 - 10 ml of Venofer (100 - 200 mg iron) 1 to 3 times a week.
For administration time and dilution ratio see "Method of administration" as follows.
Paediatric population: There is moderate amount of data in children under study conditions. If there is a clinical need, it is recommended not to exceed 0.15 ml of Venofer (3 mg iron) per kg body weight not more than three times per week.
For administration time and dilution ratio see "Method of administration" as follows.
Maximum tolerated single and weekly doses: Adults: As an injection, maximum tolerated dose per day given not more than 3 times per week: 10 ml of Venofer (200 mg iron) injected over at least 10 minutes.
As an infusion, maximum tolerated dose per day given not more than once per week: Patients above 70 kg body weight: 500 mg iron (25 ml of Venofer) over at least 3 ½ hours; Patients of 70 kg body weight and below: 7 mg iron/kg body weight over at least 3 ½ hours.
The infusion times given in "Method of administration" as follows should be strictly adhered to, even if the patient does not receive the maximum tolerated single dose.
Method of administration: Venofer must only be administered by the intravenous route. This may be by drip infusion, slow injection or directly into the venous line of the dialysis machine.
Intravenous drip infusion: Venofer must only be diluted in sterile 0.9% m/V sodium chloride (NaCl) solution. Dilution must take place immediately prior to infusion and the solution should be administered as follows: (See Table 2.)
Click on icon to see table/diagram/image
Intravenous injection: Venofer may be administered by slow intravenous injection at a rate of 1 ml undiluted solution per minute and not exceeding 10 ml (200 mg iron) per injection.
Injection into venous line of dialysis machine: Venofer may be administered during a haemodialysis session directly into the venous line of the dialysis machine under the same conditions as for intravenous injection.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image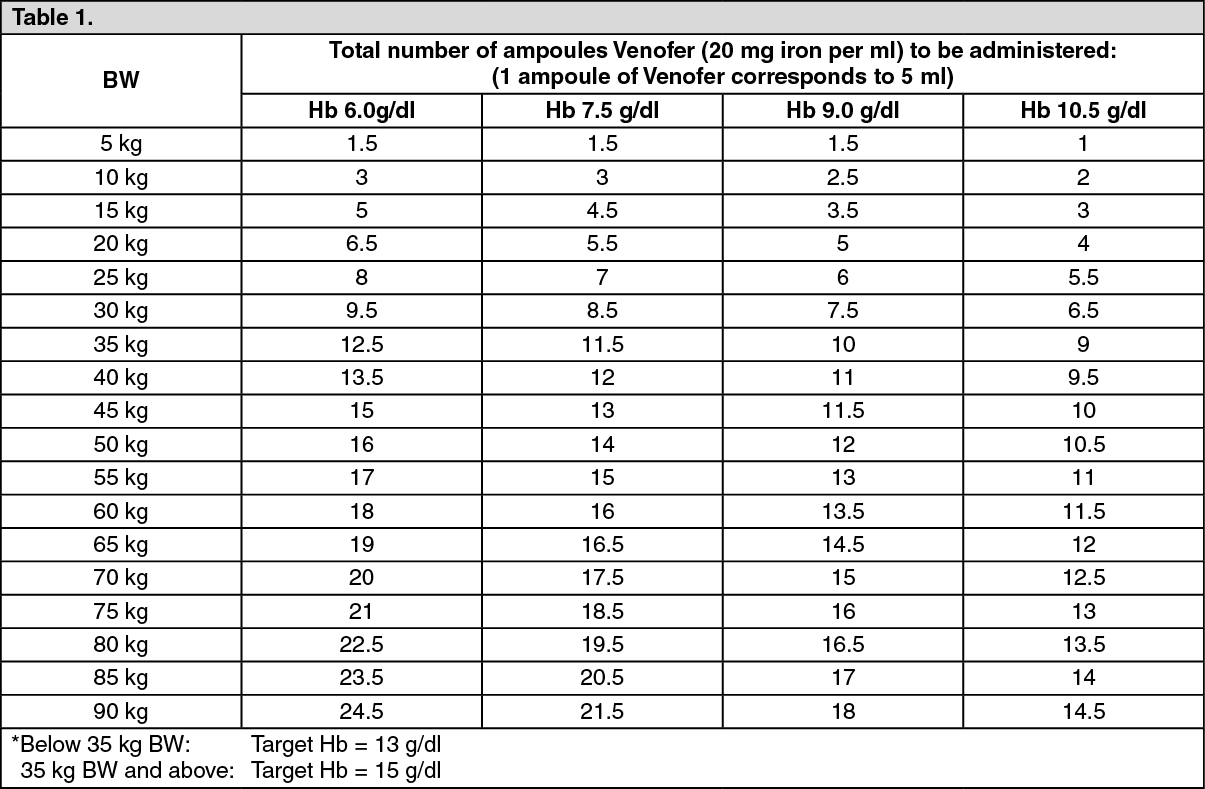 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image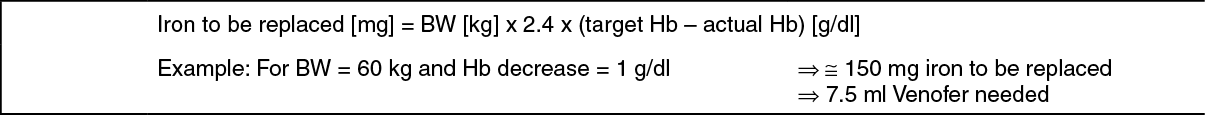 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
