WHO ATC Code: C03XA01.
Pharmacology: Mechanism of action: SAMSCA is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP). SAMSCA affinity for the V2-receptor is 29 times greater than for the V1a-receptor. When taken orally, 15 to 60 mg doses of SAMSCA antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. Urinary excretion of sodium and potassium and plasma potassium concentrations are not significantly changed. SAMSCA metabolites have no or weak antagonist activity for human V2-receptors compared with SAMSCA.
Plasma concentrations of native AVP may increase (avg. 2-9 pg/mL) with SAMSCA administration.
Pharmacodynamics: In healthy subjects receiving a single dose of SAMSCA 60 mg, the onset of the aquaretic and sodium increasing effects occurs within 2 to 4 hours post-dose. A peak effect of about a 6 mEq increase in serum sodium and about 9 mL/min increase in urine excretion rate is observed between 4 and 8 hours post-dose; thus, the pharmacological activity lags behind the plasma concentrations of SAMSCA. About 60% of the peak effect on serum sodium is sustained at 24 hours post-dose, but the urinary excretion rate is no longer elevated by this time. Doses above 60 mg SAMSCA do not increase aquaresis or serum sodium further. The effects of SAMSCA in the recommended dose range of 15 to 60 mg once daily appear to be limited to aquaresis and the resulting increase in sodium concentration.
In a parallel-arm, double-blind (for SAMSCA and placebo), placebo- and positive-controlled, multiple dose study of the effect of SAMSCA on the QTc interval, 172 healthy subjects were randomized to SAMSCA 30 mg, SAMSCA 300 mg, placebo, or moxifloxacin 400 mg once daily. At both the 30 mg and 300 mg doses, no significant effect of administering SAMSCA on the QTc interval was detected on Day 1 and Day 5. At the 300 mg dose, peak SAMSCA plasma concentrations were approximately 4-fold higher than the peak concentrations following a 30 mg dose. Moxifloxacin increased the QT interval by 12 ms at 2 hours after dosing on Day 1 and 17 ms at 1 hour after dosing on Day 5, indicating that the study was adequately designed and conducted to detect SAMSCA's effect on the QT interval, had an effect been present.
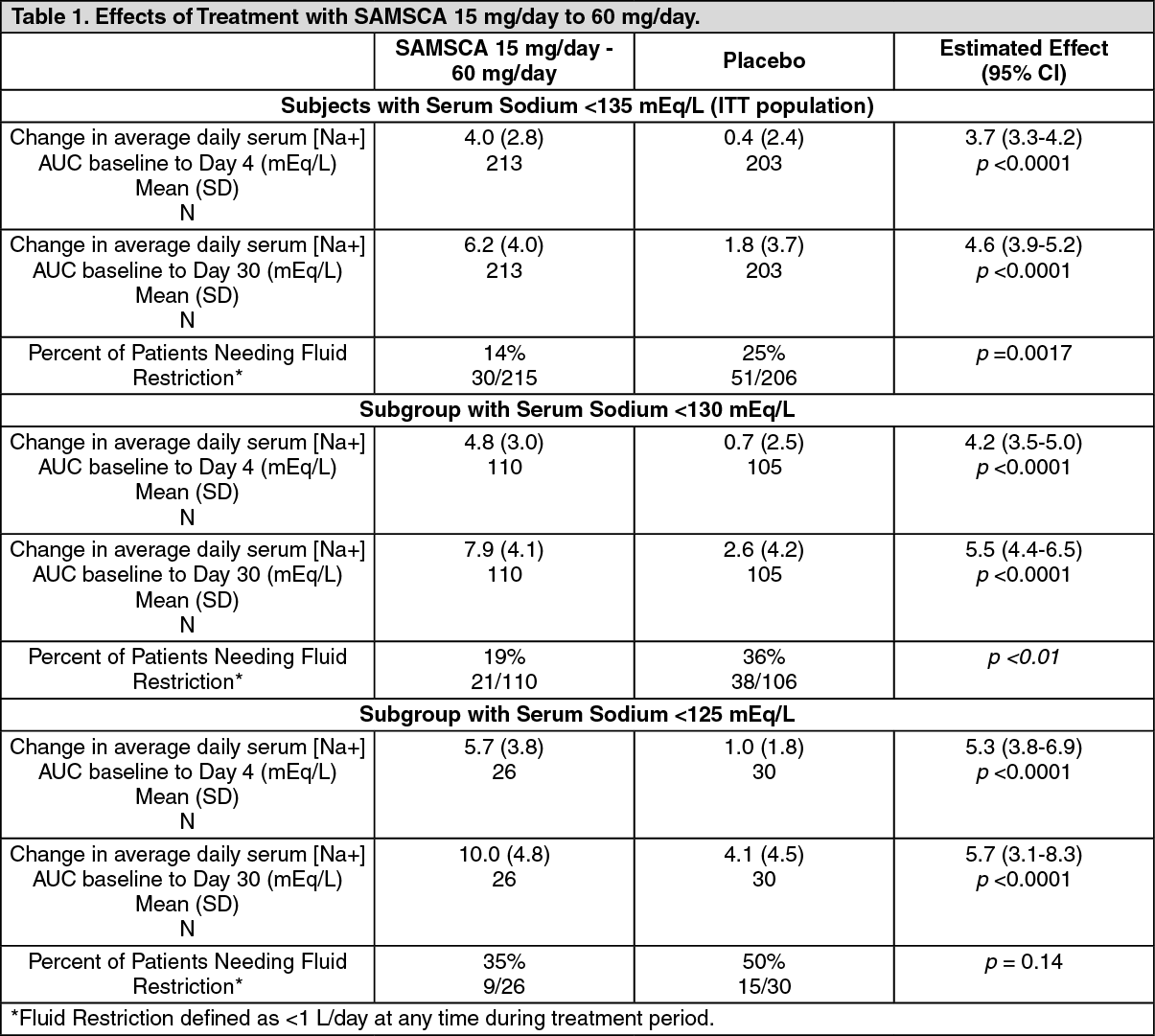
CLINICAL STUDIES: Hyponatremia: In two double-blind, placebo-controlled, multi-center studies (SALT-1 and SALT-2), a total of 424 patients with euvolemic or hypervolemic hyponatremia (serum sodium <135 mEq/L) resulting from a variety of underlying causes (heart failure, liver cirrhosis, syndrome of inappropriate antidiuretic hormone [SIADH] and others) were treated for 30 days with SAMSCA or placebo, then followed for an additional 7 days after withdrawal. Symptomatic patients, patients likely to require saline therapy during the course of therapy, patients with acute and transient hyponatremia associated with head trauma or postoperative state and patients with hyponatremia due to primary polydipsia, uncontrolled adrenal insufficiency or uncontrolled hypothyroidism were excluded. Patients were randomized to receive either placebo (N = 220) or SAMSCA (N = 223) at an initial oral dose of 15 mg once daily. The mean serum sodium concentration at study entry was 129 mEq/L. Fluid restriction was to be avoided if possible during the first 24 hours of therapy to avoid overly rapid correction of serum sodium, and during the first 24 hours of therapy 87% of patients had no fluid restriction. Thereafter, patients could resume or initiate fluid restriction (defined as daily fluid intake of ≤1.0 liter/day) as clinically indicated.The dose of SAMSCA could be increased at 24 hour intervals to 30 mg once daily, then to 60 mg once daily, until either the maximum dose of 60 mg or normonatremia (serum sodium >135 mEq/L) was reached. Serum sodium concentrations were determined at 8 hours after study drug initiation and daily up to 72 hours, within which time titration was typically completed. Treatment was maintained for 30 days with additional serum sodium assessments on Days 11, 18, 25 and 30. On the day of study discontinuation, all patients resumed previous therapies for hyponatremia and were reevaluated 7 days later. The primary endpoint for these studies was the average daily AUC for change in serum sodium from baseline to Day 4 and baseline to Day 30 in patients with a serum sodium less than 135 mEq/L. Compared to placebo, SAMSCA caused a statistically greater increase in serum sodium (p <0.0001) during both periods in both studies (see Table 1). For patients with a serum sodium of <130 mEq/L or <125 mEq/L, the effects at Day 4 and Day 30 remained significant (see Table 1). This effect was also seen across all disease etiology subsets (e.g., CHF, SIADH/other). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
SIADH/Other: For SIADH subjects in the pooled analysis, an average daily AUC of mean change from baseline in serum sodium concentration of 4.8 mEq/L for SAMSCA and 0.2 mEq/L for placebo (an estimated treatment effect of 4.7 mEq/L for SAMSCA over placebo) up to Day 4 (p < 0.0001); and of 7.4 mEq/L for SAMSCA and 1.5 mEq/L for placebo (an estimated treatment effect of 6.2 mEq/L for SAMSCA over placebo) up to Day 30 (p < 0.0001) was observed.
Congestive Heart Failure (CHF): For subjects with CHF, an average daily AUC of mean change from baseline in serum sodium concentration of 3.5 mEq/L for SAMSCA and 0.5 mEq/L for placebo (an estimated treatment effect of 3.0 mEq/L for SAMSCA over placebo) up to Day 4 (p < 0.0001); and of 6.6 mEq/L for SAMSCA and 2.4 mEq/L for placebo (an estimated treatment effect of 4.1 mEq/L for SAMSCA over placebo) up to Day 30 (p < 0.0001) was observed. Table 2 represents the pooled analysis result of the average daily AUC of mean change from baseline in serum sodium concentration by hyponatremia etiology (SIADH/other and CHF). (See Table 2.)
Click on icon to see table/diagram/image
In patients with hyponatremia (defined as <135 mEq/L), serum sodium concentration increased to a significantly greater degree in SAMSCA-treated patients compared to placebo-treated patients as early as 8 hours after the first dose, and the change was maintained for 30 days. The percentage of patients requiring fluid restriction (defined as ≤1 L/day at any time during the treatment period) was also significantly less (p =0.0017) in the SAMSCA-treated group (30/215, 14%) as compared with the placebo-treated group (51/206, 25%).
Figure 1 shows the change from baseline in serum sodium by visit in patients with serum sodium <135 mEq/L. Within 7 days of SAMSCA discontinuation, serum sodium concentrations in SAMSCA-treated patients declined to levels similar to those of placebo-treated patients. (See Figures 1 and 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the open-label study SALTWATER, 111 patients, 94 of them hyponatremic (serum sodium <135 mEq/L), previously on SAMSCA or placebo therapy were given SAMSCA as a titrated regimen (15 to 60 mg once daily) after having returned to standard care for at least 7 days. By this time, their baseline mean serum sodium concentration had fallen to between their original baseline and post-placebo therapy level. Upon initiation of therapy, average serum sodium concentrations increased to approximately the same levels as observed for those previously treated with SAMSCA, and were sustained for at least a year. Figure 3 shows results from 111 patients enrolled in the SALTWATER Study. (See Figure 3.)
Click on icon to see table/diagram/image
Adjunct treatment of volume overload in heart failure: Japanese Data: The efficacy of oral administration of SAMSCA at 15 mg or placebo once daily for 7 days in congestive heart failure patients with volume overload despite the use of a conventional diuretic was evaluated in a double-blind, placebo-controlled phase 3 study. Patients with acute heart failure were excluded. Changes in body weight from baseline at the end of treatment, the primary efficacy endpoint, were -1.54 ± 1.61 kg (mean ± SD) in the SAMSCA 15-mg group (baseline: 59.42 ± 12.30 kg, n=53) and -0.45 ± 0.93 kg in the placebo group (baseline: 55.68 ± 12.60 kg, n=57), showing that SAMSCA 15 mg significantly decreased body weight compared with placebo (p<0.0001, t-test). A marked reduction in body weight was observed in the SAMSCA 15-mg group on Day 1, with subsequent decrease during the treatment period (Figure 4). Signs and symptoms associated with cardiac edema, including jugular venous distention, hepatomegaly, and lower limb edema, were also improved at the end of treatment (Table 3). (See Figure 4 and Table 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Other data: The efficacy and safety of SAMSCA as adjunct treatment of volume overload in heart failure was evaluated in one other phase 2 study and two other phase 3 studies. In a multicenter, randomized, double-blind, placebo-controlled phase 2 study, the effect and safety of oral administration of SAMSCA at 15 mg, 30 mg, 60 mg or placebo once daily in conjunction with conventional therapy for up to 6 months on the chronic outcomes of patients with congestive heart failure (CHF) was evaluated. Of the 330 subjects receiving either 15 mg, 30 mg or 60 mg of SAMSCA, or placebo for up to 6 months, the study suggests that patients with stable CHF (NYHA Class II and III) on standard optimal background therapy did not demonstrate an improvement in outcomes when exposed to once daily dosing of SAMSCA for up to six months. However, possible additional symptomatic and outcome benefit (i.e. improvements in NYHA class and CHF symptoms such as pedal edema, jugular venous pressure (JVP), hepatomegaly, orthopnea and patent-assessed overall treatment effect) may be present in the subset of patients with evidence of congestion (edema at baseline). SAMSCA at 15mg showed some early improvement in NYHA score as early as Week 1. SAMSCA given at doses of 15 mg, 30 mg, and 60 mg once daily for up to six months was well tolerated and safe in this population.
In a randomized, double-blind, multicenter, placebo-controlled, parallel phase 3 study to evaluate the efficacy and safety of SAMSCA in the treatment of patients with cardiac edema based on the conventional therapy, patients treated with SAMSCA 15 mg once daily achieved a statistically significant greater reduction in body weight over placebo at the end of the treatment. Changes in body weight from baseline were 1.482 ± 1.947 kg (mean ± SD) in the SAMSCA 15 mg group (baseline: 67.379 ± 11.624 kg, n= 123) and 0.540 ± 1.506 kg in the placebo group (baseline: 65.480 ± 13.430 kg, n= 120), showing a statistically significant greater reduction in body weight in the SAMSCA 15 mg group compared with placebo (p<0.0001, t-test). There was a significant increase in both the improvement (SAMSCA: 85.71% vs placebo: 68.48%; p = 0.0045) and disappearance rate (SAMSCA: 68.37% vs placebo: 54.35%, p = 0.0471) of the lower extremity edema in the SAMSCA treatment group compared to the placebo group. There was also a significant difference in the disappearance rate of jugular vein engorgement in the SAMSCA group as compared to the placebo group (SAMSCA: 40.82% vs placebo: 14.89%, p = 0.0047). There was a general improvement in the other congestive symptoms such as hepatomegaly and pulmonary congestion in the SAMSCA group although the improvements did not reach statistical significance. Oral administration of SAMSCA 15 mg was safe as compared to placebo in the study.
In another multicenter, double-blind, randomized, placebo-controlled phase 3 study to evaluate the efficacy and safety of SAMSCA in the treatment of cardiac edema in patients with heart failure, the mean change in body weight from baseline were -1.36 ± 2.13 kg (mean ± SD) in the SAMSCA group and -0.59 ± 1.27 kg in the placebo group. The significant difference in the mean changes between the two groups was -0.78 kg (p=0.0394; 95% CI: -1.52 to -0.04 kg), showing a statistically significant reduction in body weight in the SAMSCA 15 mg group compared with placebo (p < 0.0001, t-test). Significant daily urine volume output was observed when compared with the placebo for the four days of treatment. Without fluid restriction, mean daily fluid intake/urine volume output balance revealed significant difference in two groups (mean daily intake/output balance: -97.5 mL in the SAMSCA group vs. 262.1 mL in the placebo group, p = 0.0131).
For pulmonary congestion, the improvement rate (SAMSCA: 47.83% vs. placebo: 42.22%, p = 0.6751) and resolution rate (SAMSCA: 29.27% vs. placebo: 25.64%; p = 0.8045) in the SAMSCA group was numerically higher than that in placebo group although no significant difference was observed between the two groups. At the end of the study, pulmonary rales remained unchanged in most subjects (SAMSCA: 60.87% vs. placebo: 68.89%) but more subjects in the SAMSCA group achieved improvement (p = 0.8386). None of the subjects showed worsened lower limb edema at the end of the study. Overall, a total of 31 subjects (67.39%) in the SAMSCA group experienced at least 1-level grade of improvement, while a total of 26 subjects in placebo showed improvement (57.78%) in the intention-to-treat (ITT) population (p = 0.3905). The severity of change from baseline in lower limb edema was compared between groups by the proportional odds model, no statistically significant difference was observed between treatment groups (OR 1.3297; p = 0.5164). For patient self-assessed dyspnea status, there was higher percentage of improvement in the dyspnea in the SAMSCA group (SAMSCA: 89.13% vs placebo: 80.00%), however this failed to meet statistical significance (OR 1.175; p = 0.7717). For physician-assessed dyspnea, 73.91% of subjects reached some grade of improvement vs. 66.67% in the placebo group, but there was no significant difference (p = 0.4971) in the mitigation of dyspnea between groups. SAMSCA was well tolerated in the study.
Pharmacokinetics: In healthy subjects the pharmacokinetics of tolvaptan after single doses of up to 480 mg and multiple doses up to 300 mg once daily have been examined. Area under the curve (AUC) increases proportionally with dose. After administration of doses ≥60 mg, however, Cmax increases less than proportionally with dose. The pharmacokinetic properties of tolvaptan are stereospecific, with a steady-state ratio of the S-(-) to the R-(+) enantiomer of about 3. The absolute bioavailability of SAMSCA is 56%. Peak concentrations of SAMSCA are observed between 2 and 4 hours post-dose. Food does not impact the bioavailability of SAMSCA.
In vitro data indicate that SAMSCA is a substrate and inhibitor of P-gp. SAMSCA is highly plasma protein bound (99%) and distributed into an apparent volume of distribution of about 3 L/kg. SAMSCA is eliminated entirely by non-renal routes and mainly, if not exclusively, metabolized by CYP 3A. After oral dosing, clearance is about 4 mL/min/kg and the terminal phase half-life is about 12 hours. The accumulation factor of SAMSCA with the once-daily regimen is 1.3 and the trough concentrations amount to ≤16% of the peak concentrations, suggesting a dominant half-life somewhat shorter than 12 hours. There is marked inter-subject variation in peak and average exposure to SAMSCA with a percent coefficient of variation ranging between 30 and 60%.
In patients with hyponatremia of any origin the clearance of SAMSCA is reduced to about 2 mL/min/kg. Moderate or severe hepatic impairment or congestive heart failure decrease the clearance and increase the volume of distribution of SAMSCA, but the respective changes are not clinically relevant. Exposure and response to SAMSCA in subjects with creatinine clearance ranging between 79 and 10 mL/min and patients with normal renal function are not different.
In a study in patients with creatinine clearances ranging from 10-124 mL/min administered a single dose of 60 mg SAMSCA, AUC and Cmax of plasma SAMSCA were less than doubled in patients with severe renal impairment relative to the controls. The peak increase in serum sodium was 5-6 mEq/L, regardless of renal function, but the onset and offset of SAMSCA's effect on serum sodium were slower in patients with severe renal impairment.
Toxicology: NONCLINICAL TOXICOLOGY: Carcinogenesis, mutagenesis, impairment of fertility
: Up to two years of oral administration of SAMSCA to male and female rats at doses up to 1000 mg/kg/day (162 times the maximum recommended human dose [MRHD] on a body surface area basis), to male mice at doses up to 60 mg/kg/day (5 times the MRHD) and to female mice at doses up to 100 mg/kg/day (8 times the MRHD) did not increase the incidence of tumors. SAMSCA tested negative for genotoxicity in
in vitro (bacterial reverse mutation assay and chromosomal aberration test in Chinese hamster lung fibroblast cells) and
in vivo (rat micronucleus assay) test systems.
In a fertility study in which male and female rats were orally administered SAMSCA at 100, 300 or 1000 mg/kg/day, the highest dose level was associated with significantly fewer corpora lutea and implants than control.
Reproductive and developmental toxicology
: In pregnant rats, oral administration of SAMSCA at 10, 100 and 1000 mg/kg/day during organogenesis was associated with a reduction in maternal body weight gain and food consumption at 100 and 1000 mg/kg/day (16 and 162 times the MRHD), and reduced fetal weight and delayed ossification of fetuses at 1000 mg/kg/day (162 times the MRHD on a body surface area basis). Oral administration of SAMSCA at 100, 300 and 1000 mg/kg/day to pregnant rabbits during organogenesis was associated with reductions in maternal body weight gain and food consumption at all doses (32 to 324 times the MRHD), and abortions at mid- and high-doses (97 to 324 times the MRHD). At 1000 mg/kg/day (324 times the MRHD), increased incidences of embryo-fetal death, fetal microphthalmia, open eyelids, cleft palate, brachymelia and skeletal malformations were observed. There are no adequate and well-controlled studies of SAMSCA in pregnant women. SAMSCA is contraindicated during pregnancy.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image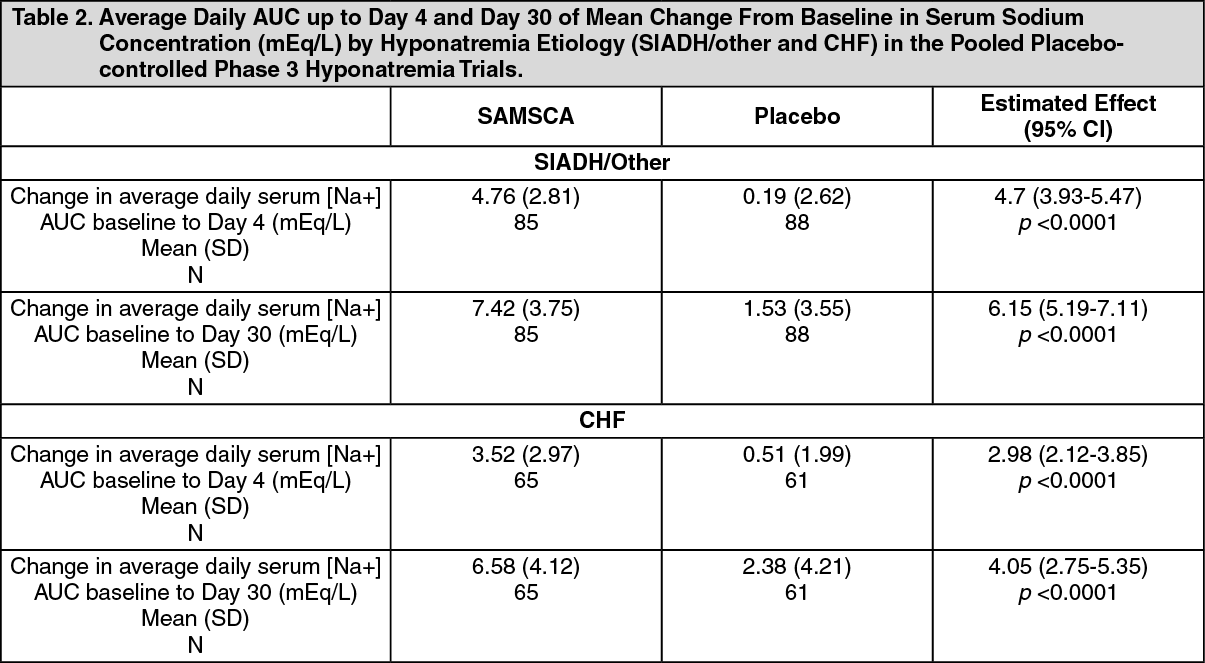 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image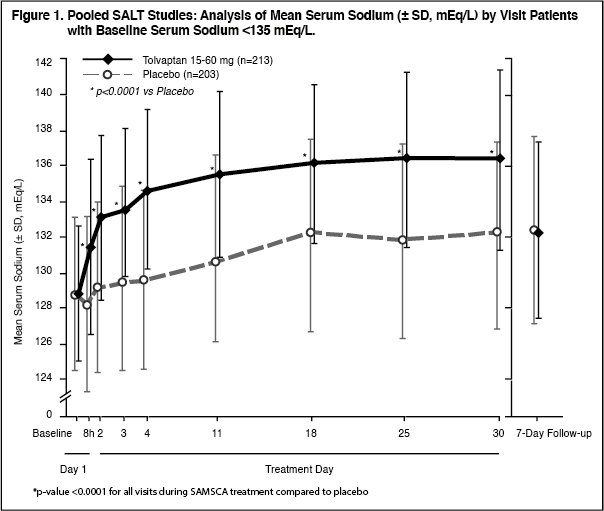 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image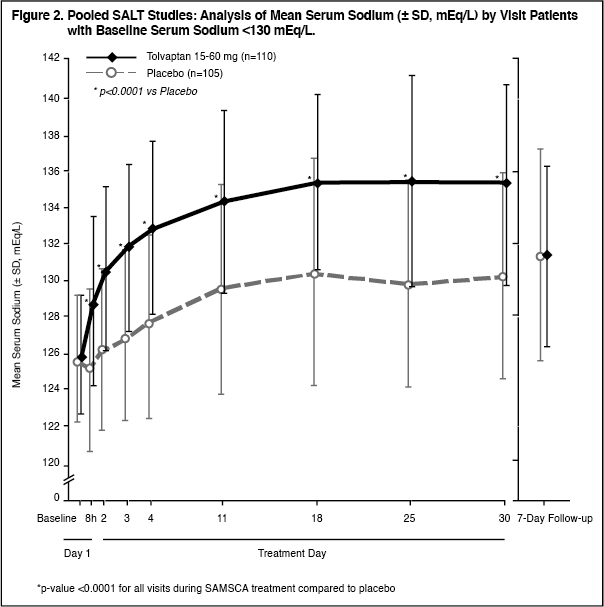 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image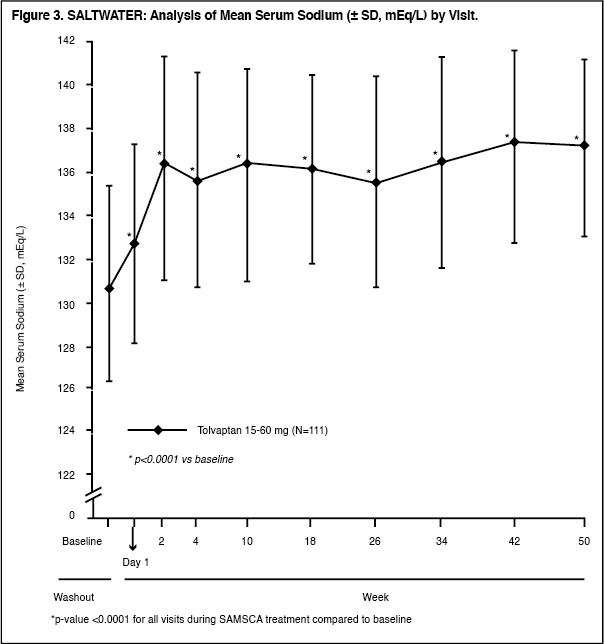 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image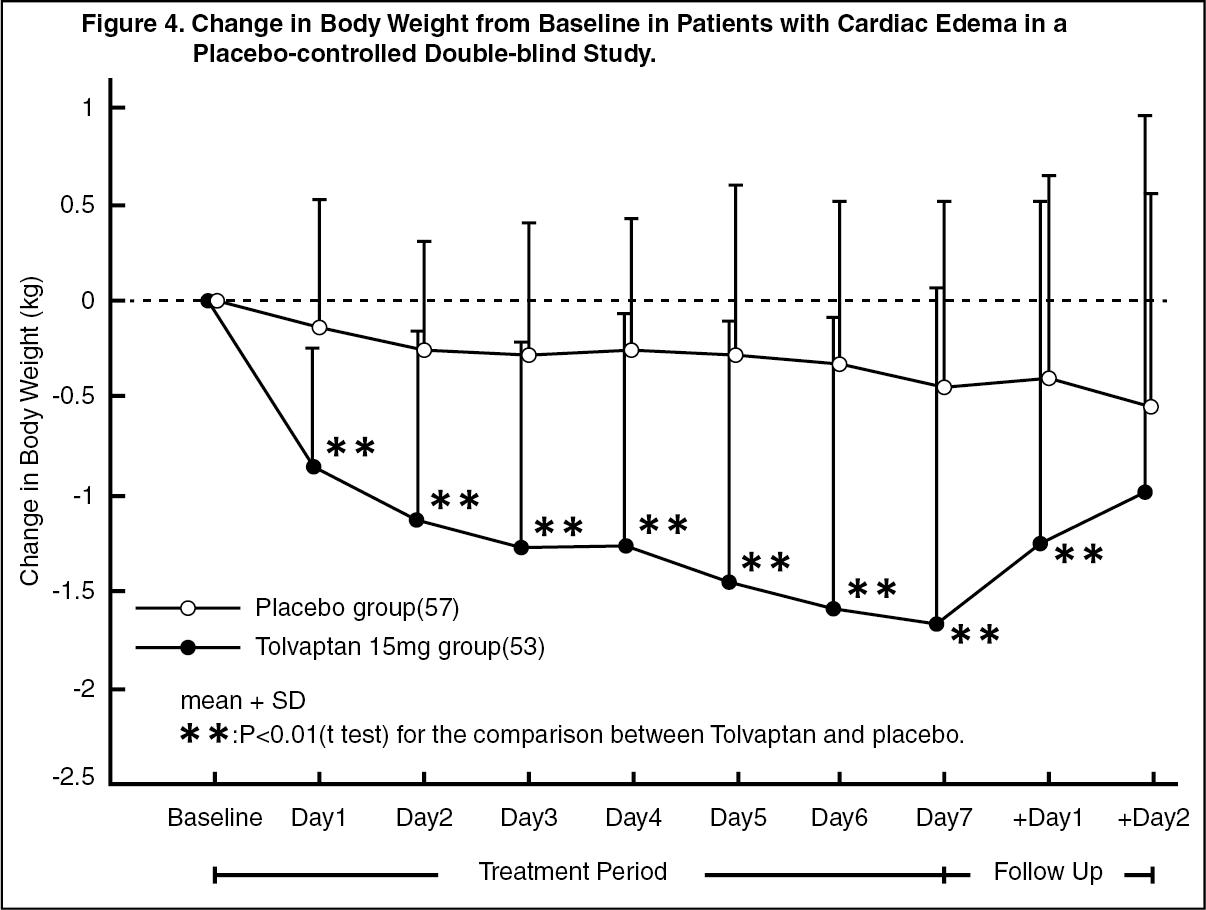 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image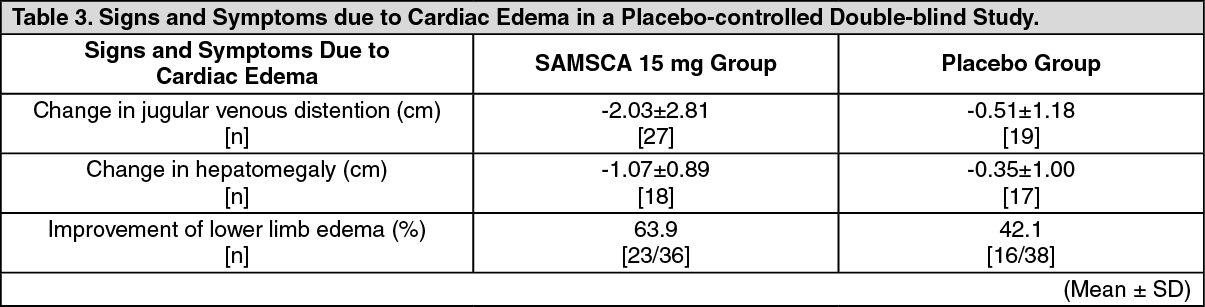 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image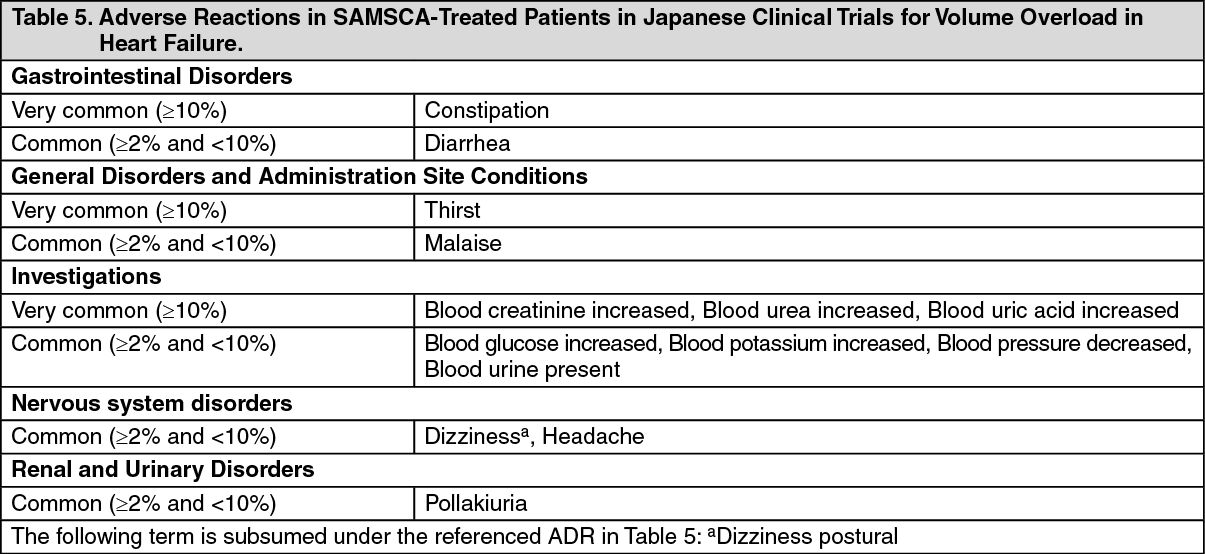 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
