Hepatic Effects: In clinical studies, persistent increases [to >3 x upper limit of normal (ULN)] in serum transaminases have occurred in a few adult patients who received simvastatin. When simvastatin was interrupted or discontinued in these patients, the transaminase levels usually fell slowly to pre-treatment levels.
If the transaminase levels show evidence of progression, particularly if they rise to 3 x ULN and are persistent, simvastatin should be discontinued.
It is recommended that liver function tests be performed before treatment begins and thereafter when clinically indicated. Patients titrated to the 80-mg dose should receive an additional test prior to titration, 3 months after titration to the 80-mg dose and periodically thereafter (eg, semi-annually) for the 1st year of treatment. Special attention should be paid to patients who develop elevated serum transaminase levels and in these patients, measurements should be repeated promptly and then performed more frequently. If the transaminase levels show evidence of progression, particularly if they rise to 3 x ULN and are persistent, simvastatin should be discontinued.
The product should be used with caution in patients who consume substantial quantities of alcohol and/or have a past history of liver disease. Active liver diseases or unexplained transaminase elevations are contraindications to the use of simvastatin.
As with other lipid-lowering agents moderate (3 x ULN) elevations of serum transaminases have been reported following therapy with simvastatin. These changes appeared soon after initiation of therapy with simvastatin, were often transient, were not accompanied by any symptoms and interruption of treatment was not required.
Myopathy/Rhabdomyolysis: Simvastatin occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase (CK) >10 x ULN. Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria and rare fatalities has occurred. The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. Predisposing factors for myopathy include advanced age (≥65 yrs), female gender, uncontrolled hypothyroidism and renal impairment.
As with other statins, the risk of myopathy/rhabdomyolysis is dose related. In a clinical trial database in which 41,413 patients were treated with simvastatin, 24,747 (approximately 60%) of whom were enrolled in studies with a median follow-up of at least 4 yrs, the incidence of myopathy was approximately 0.03%, 0.08% and 0.61% at 20, 40 and 80 mg/day, respectively. In these trials, patients were carefully monitored and some interacting medicinal products were excluded. In a clinical trial in which patients with a history of myocardial infarction were treated with simvastatin 80 mg/day (mean follow-up 6.7 yrs), the incidence of myopathy was approximately 1% compared with 0.02% for patients on 20 mg/day. Approximately half of these myopathy cases occurred during the 1st year of treatment. The incidence of myopathy during each subsequent year of treatment was approximately 0.1%. The risk of myopathy is greater in patients on simvastatin 80 mg compared with other statin-based therapies with similar LDL-C-lowering efficacy. Due to the increased risk of myopathy, including rhabdomyolysis, particularly during the 1st year of treatment, use of the 80-mg dose of simvastatin should be restricted to patients who have been taking simvastatin 80 mg chronically (eg, for ≥12 months) without evidence of muscle toxicity. Patients unable to achieve their LDL-C goal utilizing the 40-mg dose of simvastatin should not be titrated to the 80-mg dose, but should be placed on alternative LDL-C lowering treatment(s) that provides greater LDL-C lowering. In patients taking simvastatin 80 mg for whom an interacting agent is needed, a lower dose of simvastatin or an alternative statin-based regimen with less potential for drug-drug interactions should be used.
In a clinical trial in which patients at high risk of cardiovascular disease were treated with simvastatin 40 mg/day (median follow-up 3.9 yrs), the incidence of myopathy was approximately 0.05% for non-Chinese patients (n=7367) compared with 0.24% for Chinese patients (n=5468). While the only Asian population assessed in this clinical trial was Chinese, caution should be used when prescribing simvastatin to Asian patients and the lowest dose necessary should be employed.
Cases of myopathy/rhabdomyolysis have been observed with simvastatin coadministered with lipid modifying doses (>1 g/day) of niacin. In a clinical trial (median follow-up 3.9 yrs) involving patients at high risk of cardiovascular disease and with well-controlled LDL-C levels on simvastatin 40 mg/day with or without ezetimibe 10 mg, there was no incremental benefit on cardiovascular outcomes with the addition of lipid-modifying doses (≥1 g/day) of niacin. Therefore, the benefit of the combined use of simvastatin with niacin should be carefully weighed against the potential risks of the combination. In addition, in this trial, the incidence of myopathy was approximately 0.24% for Chinese patients on simvastatin 40 mg or ezetimibe/simvastatin 10/40 mg compared with 1.24% for Chinese patients on simvastatin 40 mg or ezetimibe/simvastatin 10/40 mg coadministered with extended-release niacin/laropiprant 2 g/40 mg. While the only Asian population assessed in this clinical trial was Chinese, because the incidence of myopathy is higher in Chinese than in non-Chinese patients, coadministration of simvastatin with lipid modifying doses (>1 g/day) of niacin is not recommended in Asian patients.
All patients starting therapy with simvastatin or whose dose of simvastatin is being increased, should be advised of the risk of myopathy and told to report promptly any unexplained muscle pain, tenderness or weakness. Simvastatin therapy should be discontinued immediately if myopathy is diagnosed or suspected.
The presence of these symptoms and a CK level >10 x ULN indicates myopathy. In most cases, when patients were promptly discontinued from treatment, muscle symptoms and CK increases resolved. Periodic CK determinations may be considered in patients starting therapy with simvastatin or whose dose is being increased. Periodic CK determinations are recommended for patients titrating to the 80-mg dose. There is no assurance that such monitoring will prevent myopathy.
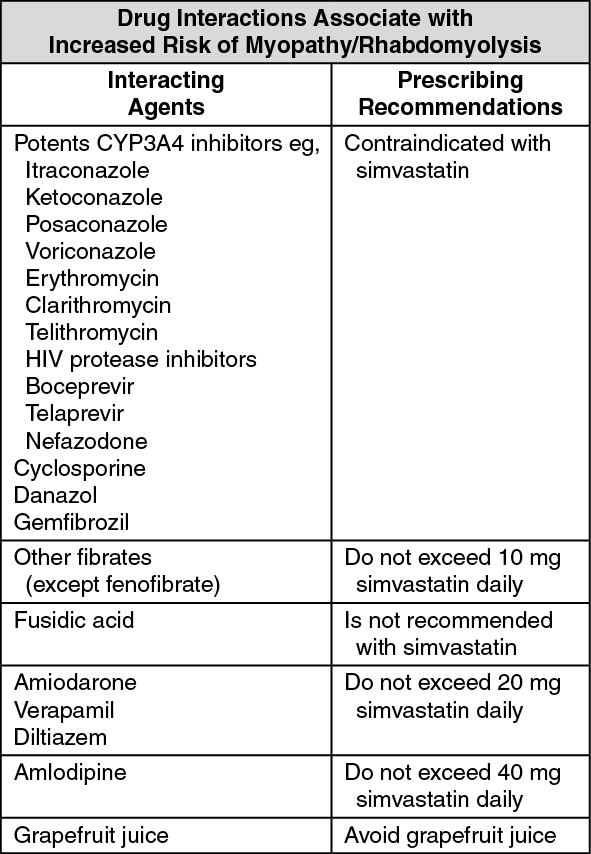
Prescribing recommendations for interacting agents are summarized in the table. (See table.)
 Click on icon to see table/diagram/image
Creatine Kinase Measurement:
Click on icon to see table/diagram/image
Creatine Kinase Measurement: Creatine Kinase (CK) levels should not be measured following strenuous exercise or in the presence of any plausible alternative cause of CK increase as this makes value interpretation difficult. If CK levels are significantly elevated at baseline (>5 x ULN), levels should be re-measured within 5-7 days later to confirm the results.
Before the Treatment: All patients starting therapy with simvastatin or whose dose of simvastatin is being increased, should be advised of the risk of myopathy and told to report promptly any unexplained muscle pain, tenderness or weakness.
Caution should be exercised in patients with predisposing factors for rhabdomyolysis. In order to establish a reference baseline value, CK level should be measured before starting treatment in the following situations: Elderly (>65 years); female gender; renal impairment; uncontrolled hypothyroidism; personal or familial history of hereditary muscular disorders; previous history of muscular toxicity with a statin or fibrate; alcohol abuse.
In such situations, the risk of treatment should be considered in relation to possible benefit and clinical monitoring is recommended. If a patient has previously experienced a muscle disorder on a fibrate or a statin treatment with a different member of the class should only be initiated with caution. If CK levels are significantly elevated at baseline (>5 x ULN), treatment should not be started.
While on Treatment: If muscular pain, weakness or cramps occur whilst a patient is receiving treatment with a statin, their CK levels should be measured. If these levels are found, in the absence of strenuous exercise, to be significantly elevated (>5 x ULN), treatment should be stopped. If muscular symptoms are severe and cause daily discomfort, even if CK levels are <5 x ULN, treatment discontinuation may be considered. If myopathy is suspected for any other reason, treatment should be discontinued.
If symptoms resolve and CK levels return to normal then reintroduction of the statin or introduction of an alternative statin may be considered at the lowest dose and with close monitoring.
Therapy with simvastatin should be temporarily stopped a few days prior to elective major surgery and when any major medical or surgical condition supervenes.
Measures to Reduce the Risk of Myopathy Caused by Medicinal Product Interactions (see Interactions): The risk of myopathy and rhabdomyolysis is significantly increased by concomitant use of simvastatin with potent inhibitors of CYP3A4 (eg, itraconazole, ketoconazole, erythromycin, clarithromycin, telithromycin, HIV protease inhibitors, nefazodone), as well as gemfibrozil, ciclosporin and danazol (see Dosage & Administration).
The risk of myopathy and rhabdomyolysis is also increased by concomitant use of other fibrates, lipid-lowering doses of niacin (≥1 g/day) or by concomitant use of amiodarone or verapamil with higher doses of simvastatin (see Dosage & Administration and Interactions). There is also a slight increase in risk when diltiazem or amlodipine is used with simvastatin 80 mg. The risk of myopathy including rhabdomyolysis may be increased by concomitant administration of fusidic acid with statins (see Interactions).
Consequently, regarding CYP3A4 inhibitors, the use of simvastatin concomitantly with itraconazole, ketoconazole, HIV protease inhibitors, erythromycin, clarithromycin, telithromycin and nefazodone is contraindicated (see Contraindications and Interactions). If treatment with itraconazole, ketoconazole, erythromycin, clarithromycin or telithromycin is unavoidable, therapy with simvastatin must be suspended during the course of treatment.
Moreover, caution should be exercised when combining simvastatin with certain other less potent CYP3A4 inhibitors: Ciclosporin, verapamil, diltiazem (see Dosage & Administration and Interactions).
Concomitant intake of grapefruit juice and simvastatin should be avoided.
The dose of simvastatin should not exceed 10 mg daily in patients receiving concomitant medication with ciclosporin, danazol, gemfibrozil or lipid-lowering doses (≥1 g/day) of niacin. The combined use of simvastatin with gemfibrozil should be avoided, unless the benefits are likely to outweigh the increased risks of this drug combination. The benefits of the combined use of simvastatin 10 mg daily with other fibrates (except fenofibrate), niacin, ciclosporin or danazol should be carefully weighed against the potential risks of these combinations. (See Dosage & Administration and Interactions.)
Caution should be used when prescribing fenofibrate with simvastatin, as either agent can cause myopathy when given alone.
The combined use of simvastatin at doses >20 mg daily with amiodarone or verapamil should be avoided unless the clinical benefit is likely to outweigh the increased risk of myopathy (see Dosage & Administration and Interactions).
In an interim analysis of an ongoing clinical outcomes study, an independent safety monitoring committee identified a higher than expected incidence of myopathy in Chinese patients taking simvastatin 40 mg and nicotinic acid/laropiprant 2000 mg/40 mg. Therefore, caution should be used when treating Chinese patients with simvastatin (particularly doses of ≥40 mg) co-administered with lipid-modifying doses niacin (nicotinic acid ≥1 g/day) or products containing niacin. Because the risk of myopathy with statins is dose-related, the use of simvastatin 80 mg with lipid-modifying doses niacin (nicotinic acid ≥1 g/day) or products containing niacin is not recommended in Chinese patients. It is unknown whether there is an increased risk of myopathy in other Asian patients treated with simvastatin co-administered with lipid-modifying doses niacin (nicotinic acid ≥1 g/day) or products containing niacin.
If the combination proves necessary, patients on fusidic acid and simvastatin should be closely monitored (see Interactions). Temporary suspension of simvastatin treatment may be considered.
Excipient: Patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Effects on the Ability to Drive or Operate Machinery: Priacin has no or negligible influence on the ability to drive and use machines.
However, when driving vehicles or operating machines, it should be taken into account that dizziness has been reported rarely in post-marketing experiences.
Use in pregnancy: Safety in pregnant women has not been established. No controlled clinical trials with simvastatin have been conducted in pregnant women. Rare reports of congenital anomalies following intrauterine exposure to HMG-CoA reductase inhibitors have been received. However, in an analysis of approximately 200 prospectively followed pregnancies exposed during the 1st trimester to Priacin or another closely related HMG-CoA reductase inhibitor, the incidence of congenital anomalies was comparable to that seen in the general population. This number of pregnancies was statistically sufficient to exclude a ≥2.5-fold increase in congenital anomalies over the background incidence.
Although there is no evidence that the incidence of congenital anomalies in offspring of patients taking Priacin or another closely related HMG-CoA reductase inhibitor differs from that observed in the general population, maternal treatment with Priacin may reduce the foetal levels of mevalonate which is a precursor of cholesterol biosynthesis. Atherosclerosis is a chronic process and ordinarily discontinuation of lipid-lowering medicinal products during pregnancy should have little impact on the long-term risk associated with primary hypercholesterolaemia. For these reasons, Priacin should not be used in women who are pregnant, trying to become pregnant or suspect they are pregnant. Treatment with Priacin should be suspended for the duration of pregnancy or until it has been determined that the woman is not pregnant.
Use in lactation: It is not known whether simvastatin or its metabolites are excreted in human milk. Because many medicinal products are excreted in human milk and because of the potential for serious adverse reactions, women taking Priacin should not breastfeed their infants.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
