


 Sign Out
Sign Out



Clinical Studies: The safety and efficacy of entecavir were evaluated in three Phase 3 active-controlled trials [see Outcomes at 48 Weeks and Outcomes beyond 48 Weeks as follows]. These studies included 1633 subjects 16 years of age or older with chronic hepatitis B virus infection (serum HBsAg-positive for at least 6 months) accompanied by evidence of viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR assay). Subjects had persistently elevated ALT levels at least 1.3 times ULN and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral hepatitis. The safety and efficacy of entecavir were also evaluated in a study of 191 HBV infected subjects with decompensated liver disease and in a study of 68 subjects co-infected with HBV and HIV [see Outcomes at 48 Weeks as follows].
Outcomes at 48 Weeks: Nucleoside-naïve Subjects with Compensated Liver Disease: HBeAg-positive: Study AI463022 was a multinational, randomized, double-blind study of entecavir 0.5 mg once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 709 (of 715 randomized) nucleoside-naïve subjects with chronic hepatitis B virus infection, compensated liver disease and detectable HBeAg. The mean age of subjects was 35 years, 75% were male, 57% were Asian, 40% were Caucasian, and 13% had previously received interferon-α. At baseline, subjects had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 9.66 log10 copies/mL, and mean serum ALT level was 143 U/L. Paired, adequate liver biopsy samples were available for 89% of subjects.
HBeAg-negative (anti-HBe-positive/HBV DNA-positive): Study AI463027 was a multinational, randomized, double-blind study of entecavir 0.5 mg once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 638 (of 648 randomized) nucleoside-naïve subjects with HBeAg-negative (HBeAb-positive) chronic hepatitis B virus infection and compensated liver disease. The mean age of subjects was 44 years, 76% were male, 39% were Asian, 58% were Caucasian, and 13% had previously received interferon-α. At baseline, subjects had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 7.58 log10 copies/mL, and mean serum ALT level was 142 U/L. Paired, adequate liver biopsy samples were available for 88% of subjects.
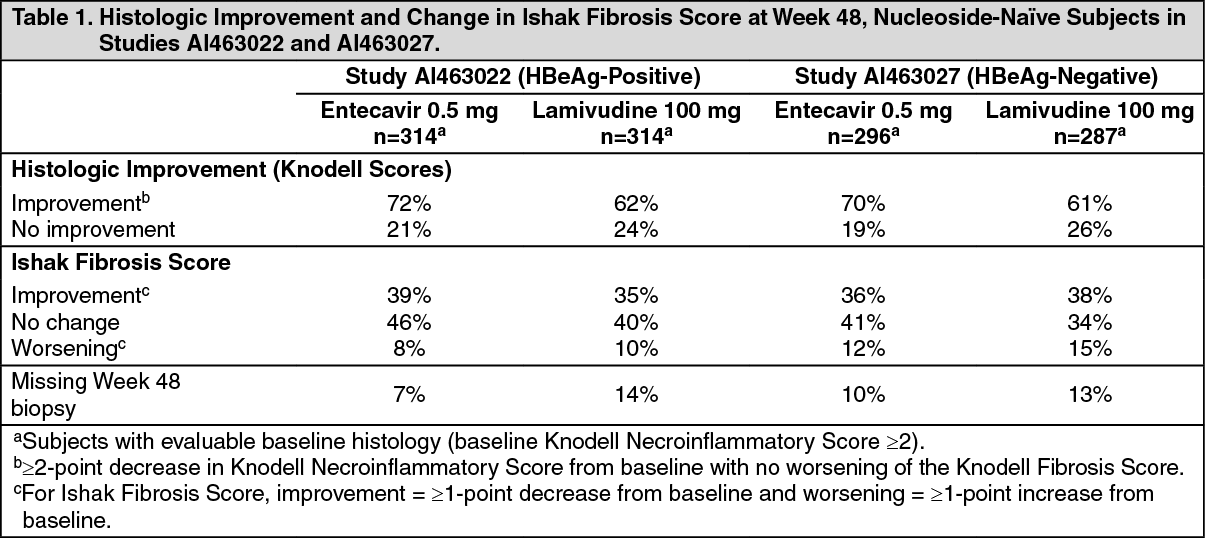
In Studies AI463022 and AI463027, entecavir was superior to lamivudine on the primary efficacy endpoint of Histologic Improvement, defined as a 2-point or greater reduction in Knodell Necroinflammatory Score with no worsening in Knodell Fibrosis Score at Week 48, and on the secondary efficacy measures of reduction in viral load and ALT normalization. Histologic Improvement and change in Ishak Fibrosis Score are shown in Table 1. Selected virologic, biochemical, and serologic outcome measures are shown in Table 2. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image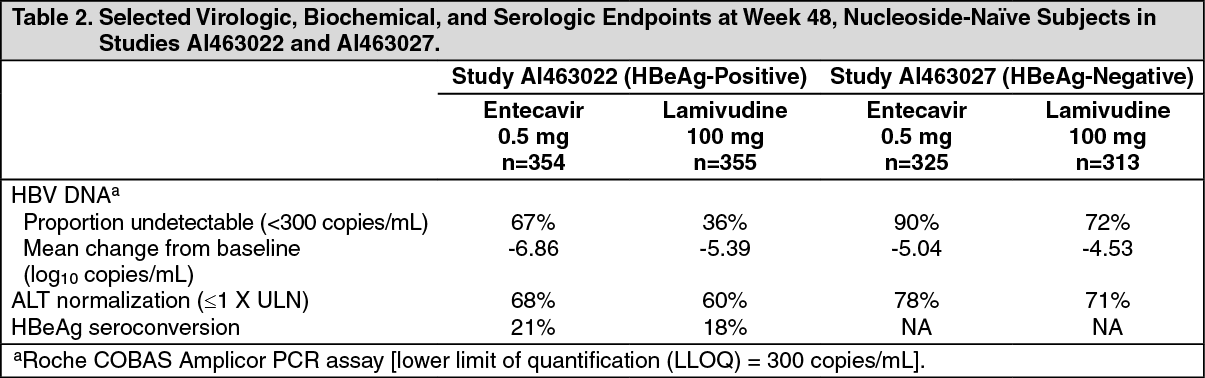 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHistologic Improvement was independent of baseline levels of HBV DNA or ALT.
Lamivudine-refractory Subjects with Compensated Liver Disease: Study AI463026 was a multinational, randomized, double-blind study of entecavir in 286 (of 293 randomized) subjects with lamivudine-refractory chronic hepatitis B virus infection and compensated liver disease. Subjects receiving lamivudine at study entry either switched to entecavir 1 mg once daily (with neither a washout nor an overlap period) or continued on lamivudine 100 mg for a minimum of 52 weeks. The mean age of subjects was 39 years, 76% were male, 37% were Asian, 62% were Caucasian, and 52% had previously received interferon-α. The mean duration of prior lamivudine therapy was 2.7 years, and 85% had lamivudine resistance mutations at baseline by an investigational line probe assay. At baseline, subjects had a mean Knodell Necroinflammatory Score of 6.5, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 9.36 log10 copies/mL, and mean serum ALT level was 128 U/L. Paired, adequate liver biopsy samples were available for 87% of subjects.
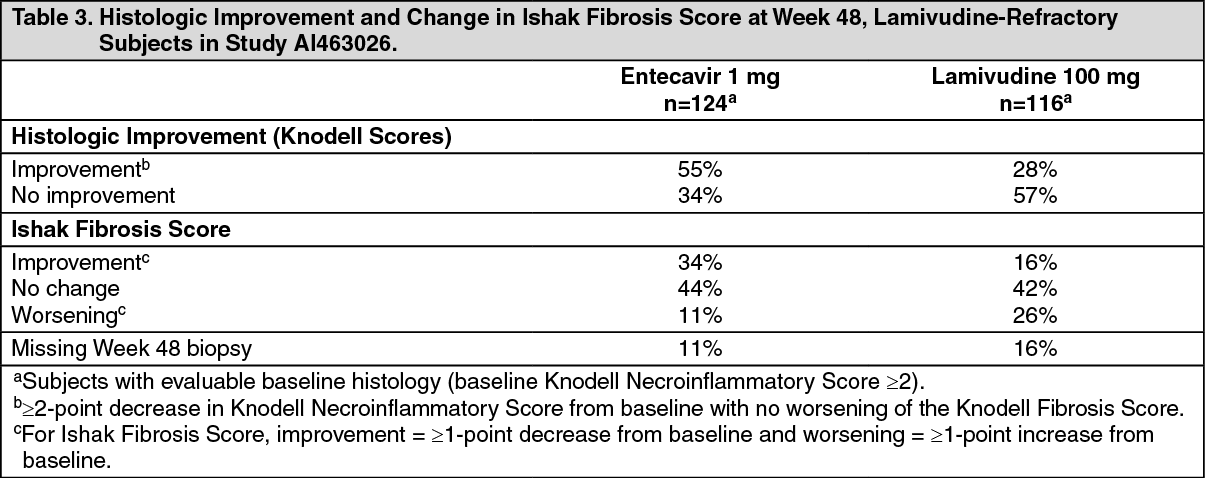
Entecavir was superior to lamivudine on a primary endpoint of Histologic Improvement (using the Knodell Score at Week 48). These results and change in Ishak Fibrosis Score are shown in Table 3. Table 4 shows selected virologic, biochemical, and serologic endpoints. (See Tables 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image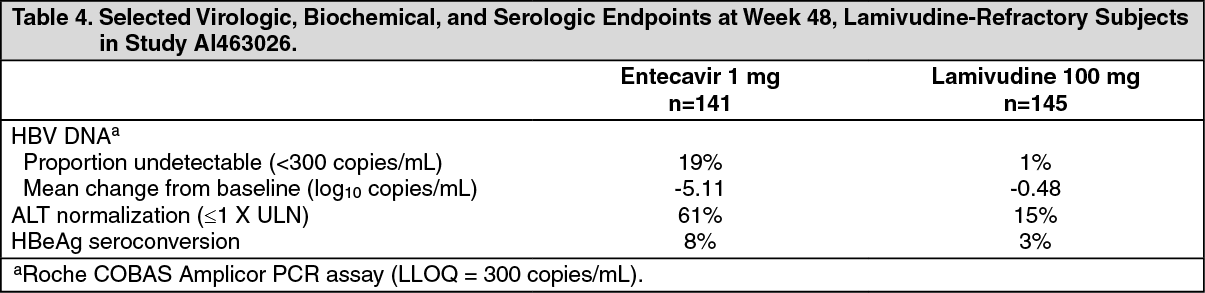 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHistologic Improvement was independent of baseline levels of HBV DNA or ALT.
Subjects with Decompensated Liver Disease: Study AI463048 was a randomized, open-label study of entecavir 1 mg once daily versus adefovir dipivoxil 10 mg once daily in 191 (of 195 randomized) adult subjects with HBeAg-positive or -negative chronic HBV infection and evidence of hepatic decompensation, defined as a Child-Turcotte-Pugh (CTP) score of 7 or higher. Subjects were either HBV-treatment-naïve or previously treated, predominantly with lamivudine or interferon-α.
In Study AI463048, 100 subjects were randomized to treatment with entecavir and 91 subjects to treatment with adefovir dipivoxil. Two subjects randomized to treatment with adefovir dipivoxil actually received treatment with entecavir for the duration of the study. The mean age of subjects was 52 years, 74% were male, 54% were Asian, 33% were Caucasian, and 5% were Black/African American. At baseline, subjects had a mean serum HBV DNA by PCR of 7.83 log10 copies/mL and mean ALT level of 100 U/L; 54% of subjects were HBeAg-positive; 35% had genotypic evidence of lamivudine resistance. The baseline mean CTP score was 8.6. Results for selected study endpoints at Week 48 are shown in Table 5. (See Table 5.)
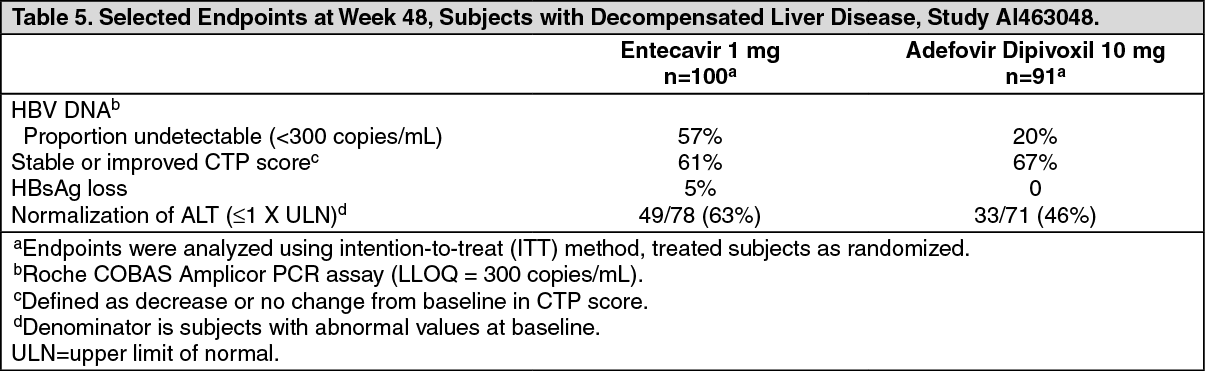 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSubjects Co-infected with HIV and HBV: Study AI463038 was a randomized, double-blind, placebo-controlled study of entecavir versus placebo in 68 subjects co-infected with HIV and HBV who experienced recurrence of HBV viremia while receiving a lamivudine-containing highly active antiretroviral (HAART) regimen. Subjects continued their lamivudine-containing HAART regimen (lamivudine dose 300 mg/day) and were assigned to add either entecavir 1 mg once daily (51 subjects) or placebo (17 subjects) for 24 weeks followed by an open-label phase for an additional 24 weeks where all subjects received entecavir . At baseline, subjects had a mean serum HBV DNA level by PCR of 9.13 log10 copies/mL. Ninety-nine percent of subjects were HBeAg-positive at baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable at approximately 2 log10 copies/mL through 24 weeks of blinded therapy. Virologic and biochemical endpoints at Week 24 are shown in Table 6. There are no data in patients with HIV/HBV co-infection who have not received prior lamivudine therapy. Entecavir has not been evaluated in HIV/HBV co-infected patients who were not simultaneously receiving effective HIV treatment [see Patients Co-infected with HIV and HBV under Precautions]. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor subjects originally assigned to entecavir, at the end of the open-label phase (Week 48), 8% of subjects had HBV DNA <300 copies/mL by PCR, the mean change from baseline HBV DNA by PCR was -4.20 log10 copies/mL, and 37% of subjects with abnormal ALT at baseline had ALT normalization (≤1 X ULN).
Outcomes beyond 48 Weeks: The optimal duration of therapy with entecavir is unknown. According to protocol-mandated criteria in the Phase 3 clinical trials, subjects discontinued entecavir or lamivudine treatment after 52 weeks according to a definition of response based on HBV virologic suppression (<0.7 MEq/mL by bDNA assay) and loss of HBeAg (in HBeAg-positive subjects) or ALT <1.25 X ULN (in HBeAg-negative subjects) at Week 48. Subjects who achieved virologic suppression but did not have serologic response (HBeAg-positive) or did not achieve ALT <1.25 X ULN (HBeAg-negative) continued blinded dosing through 96 weeks or until the response criteria were met. These protocol-specified subject management guidelines are not intended as guidance for clinical practice.
Nucleoside-naïve subjects: Among nucleoside-naïve, HBeAg-positive subjects (Study AI463022), 243 (69%) entecavir-treated subjects and 164 (46%) lamivudine-treated subjects continued blinded treatment for up to 96 weeks. Of those continuing blinded treatment in Year 2, 180 (74%) entecavir subjects and 60 (37%) lamivudine subjects achieved HBV DNA <300 copies/mL by PCR at the end of dosing (up to 96 weeks). 193 (79%) entecavir subjects achieved ALT ≤1 X ULN compared to 112 (68%) lamivudine subjects, and HBeAg seroconversion occurred in 26 (11%) entecavir subjects and 20 (12%) lamivudine subjects.
Among nucleoside-naïve, HBeAg-positive subjects, 74 (21%) entecavir subjects and 67 (19%) lamivudine subjects met the definition of response at Week 48, discontinued study drugs, and were followed off treatment for 24 weeks. Among entecavir responders, 26 (35%) subjects had HBV DNA <300 copies/mL, 55 (74%) subjects had ALT ≤1 X ULN, and 56 (76%) subjects sustained HBeAg seroconversion at the end of follow-up. Among lamivudine responders, 20 (30%) subjects had HBV DNA <300 copies/mL, 41 (61%) subjects had ALT ≤1 X ULN, and 47 (70%) subjects sustained HBeAg seroconversion at the end of follow-up.
Among nucleoside-naïve, HBeAg-negative subjects (Study AI463027), 26 (8%) entecavir-treated subjects and 28 (9%) lamivudine-treated subjects continued blinded treatment for up to 96 weeks. In this small cohort continuing treatment in Year 2, 22 entecavir and 16 lamivudine subjects had HBV DNA <300 copies/mL by PCR, and 7 and 6 subjects, respectively, had ALT ≤1 X ULN at the end of dosing (up to 96 weeks).
Among nucleoside-naïve, HBeAg-negative subjects, 275 (85%) entecavir subjects and 245 (78%) lamivudine subjects met the definition of response at Week 48, discontinued study drugs, and were followed off treatment for 24 weeks. In this cohort, very few subjects in each treatment arm had HBV DNA <300 copies/mL by PCR at the end of follow-up. At the end of follow-up, 126 (46%) entecavir subjects and 84 (34%) lamivudine subjects had ALT ≤1 X ULN.
Liver biopsy results: 57 subjects from the pivotal nucleoside-naïve Studies AI463022 (HBeAg-positive) and AI463027 (HBeAg-negative) who enrolled in a long-term rollover study were evaluated for long-term liver histology outcomes. The entecavir dosage was 0.5 mg daily in the pivotal studies (mean exposure 85 weeks) and 1 mg daily in the rollover study (mean exposure 177 weeks), and 51 subjects in the rollover study initially also received lamivudine (median duration 29 weeks). Of these subjects 55 (96%) had histological improvement as previously defined (see Table 3, footnote b), and 50 (88%) had a ≥1-point decrease in Ishak fibrosis score. For the 43 subjects with baseline Ishak Fibrosis Score ≥2, 25 (58%) had a ≥2-point decrease. All 10 subjects with advanced fibrosis or cirrhosis at baseline (Ishak Fibrosis Score of 4, 5 or 6) had a ≥1 point decrease (median decrease from baseline was 1.5 points). At the time of the long-term biopsy, all subjects had HBV DNA < 300 copies/mL and 49 (86%) had serum ALT ≤1 x ULN. All 57 subjects remained positive for HBsAg.
Lamivudine-refractory subjects: Among lamivudine-refractory subjects (Study AI463026), 77 (55%) entecavir-treated subjects and 3 (2%) lamivudine subjects continued blinded treatment for up to 96 weeks. In this cohort of entecavir subjects, 31 (40%) subjects achieved HBV DNA <300 copies/mL, 62 (81%) subjects had ALT ≤1 X ULN, and 8 (10%) subjects demonstrated HBeAg seroconversion at the end of dosing.
Pharmacokinetics: The single- and multiple-dose pharmacokinetics of entecavir were evaluated in healthy subjects and subjects with chronic hepatitis B virus infection.
Absorption: Following oral administration in healthy subjects, entecavir peak plasma concentrations occurred between 0.5 and 1.5 hours. Following multiple daily doses ranging from 0.1 to 1.0 mg, Cmax and area under the concentration-time curve (AUC) at steady state increased in proportion to dose. Steady state was achieved after 6 to 10 days of once-daily administration with approximately 2-fold accumulation. For a 0.5-mg oral dose, Cmax at steady state was 4.2 ng/mL and trough plasma concentration (Ctrough) was 0.3 ng/mL. For a 1 mg oral dose, Cmax was 8.2 ng/mL and Ctrough was 0.5 ng/mL.
In healthy subjects, the bioavailability of the tablet was 100% relative to the oral solution. The oral solution and tablet may be used interchangeably.
Effects of food on oral absorption: Oral administration of 0.5 mg of entecavir with a standard high-fat meal (945 kcal, 54.6 g fat) or a light meal (379 kcal, 8.2 g fat) resulted in a delay in absorption (1.0-1.5 hours fed vs. 0.75 hours fasted), a decrease in Cmax of 44%-46%, and a decrease in AUC of 18%-20% [see Dosage & Administration].
Distribution: Based on the pharmacokinetic profile of entecavir after oral dosing, the estimated apparent volume of distribution is in excess of total body water, suggesting that entecavir is extensively distributed into tissues.
Binding of entecavir to human serum proteins in vitro was approximately 13%.
Metabolism and Elimination: Following administration of 14C-entecavir in humans and rats, no oxidative or acetylated metabolites were observed. Minor amounts of phase II metabolites (glucuronide and sulfate conjugates) were observed. Entecavir is not a substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system [see Interactions as follows].
After reaching peak concentration, entecavir plasma concentrations decreased in a bi-exponential manner with a terminal elimination half-life of approximately 128-149 hours. The observed drug accumulation index is approximately 2-fold with once-daily dosing, suggesting an effective accumulation half-life of approximately 24 hours.
Entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at steady state ranging from 62% to 73% of the administered dose. Renal clearance is independent of dose and ranges from 360 to 471 mL/min suggesting that entecavir undergoes both glomerular filtration and net tubular secretion [see Interactions].
Special Populations: Gender: There are no significant gender differences in entecavir pharmacokinetics.
Race: There are no significant racial differences in entecavir pharmacokinetics.
Elderly: The effect of age on the pharmacokinetics of entecavir was evaluated following administration of a single 1 mg oral dose in healthy young and elderly volunteers. Entecavir AUC was 29.3% greater in elderly subjects compared to young subjects. The disparity in exposure between elderly and young subjects was most likely attributable to differences in renal function. Dosage adjustment of pms-ENTECAVIR should be based on the renal function of the patient, rather than age [see Renal Impairment under Dosage & Administration].
Pediatrics: Pharmacokinetic studies have not been conducted in children.
Renal impairment: The pharmacokinetics of entecavir following a single 1-mg dose were studied in subjects (without chronic hepatitis B virus infection) with selected degrees of renal impairment, including subjects whose renal impairment was managed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD). Results are shown in Table 7 [see Renal Impairment under Dosage & Administration]. (See Table 7.)
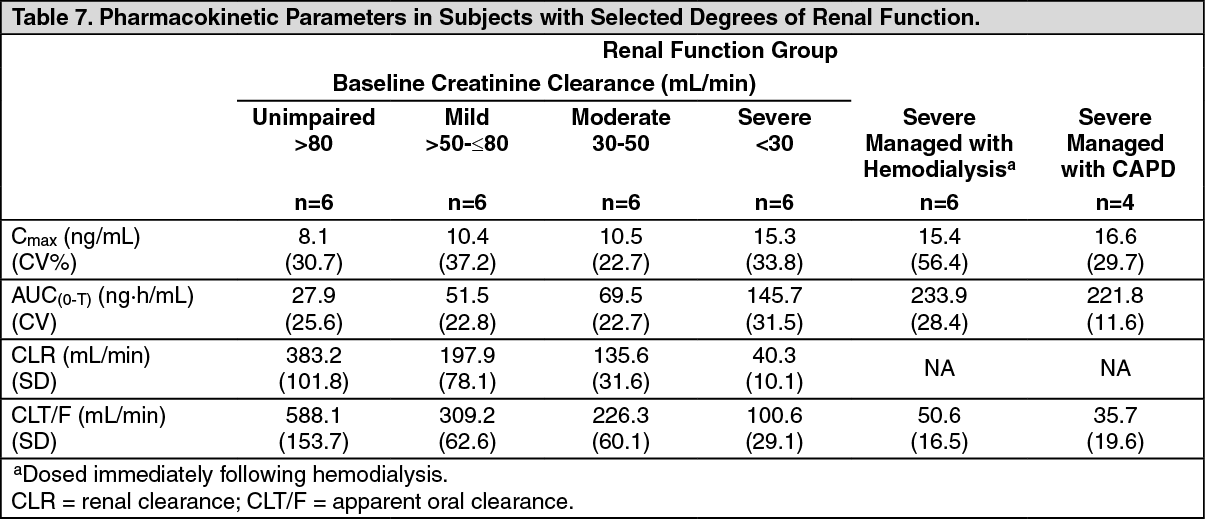 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFollowing a single 1-mg dose of entecavir administered 2 hours before the hemodialysis session, hemodialysis removed approximately 13% of the entecavir dose over 4 hours. CAPD removed approximately 0.3% of the dose over 7 days [see Renal Impairment under Dosage & Administration].
Hepatic impairment: The pharmacokinetics of entecavir following a single 1 mg dose were studied in subjects (without chronic hepatitis B virus infection) with moderate or severe hepatic impairment (Child-Turcotte-Pugh Class B or C). The pharmacokinetics of entecavir were similar between hepatically impaired and healthy control subjects; therefore, no dosage adjustment of pms-ENTECAVIR is recommended for patients with hepatic impairment.
Post-liver transplant: Limited data are available on the safety and efficacy of entecavir in liver transplant recipients. In a small pilot study of entecavir use in HBV-infected liver transplant recipients on a stable dose of cyclosporine A (n=5) or tacrolimus (n=4), entecavir exposure was approximately 2-fold the exposure in healthy subjects with normal renal function. Altered renal function contributed to the increase in entecavir exposure in these subjects. The potential for pharmacokinetic interactions between entecavir and cyclosporine A or tacrolimus was not formally evaluated [see Use in Specific Populations: Liver Transplant Recipients under Precautions].
Drug Interactions: The metabolism of entecavir was evaluated in in vitro and in vivo studies. Entecavir is not a substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. At concentrations up to approximately 10,000-fold higher than those obtained in humans, entecavir inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6, and 2E1. At concentrations up to approximately 340-fold higher than those observed in humans, entecavir did not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5, and 2B6. The pharmacokinetics of entecavir are unlikely to be affected by coadministration with agents that are either metabolized by, inhibit, or induce the CYP450 system. Likewise, the pharmacokinetics of known CYP substrates are unlikely to be affected by coadministration of entecavir.
The steady-state pharmacokinetics of entecavir and coadministered drug were not altered in interaction studies of entecavir with lamivudine, adefovir dipivoxil, and tenofovir disoproxil fumarate [see Interactions].
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Long-term oral carcinogenicity studies of entecavir in mice and rats were carried out at exposures up to approximately 42 times (mice) and 35 times (rats) those observed in humans at the highest recommended dose of 1 mg/day. In mouse and rat studies, entecavir was positive for carcinogenic findings. It is not known how predictive the results of rodent carcinogenicity studies may be for humans [see Postmarketing Experience under Adverse Reactions].
In mice, lung adenomas were increased in males and females at exposures 3 and 40 times those in humans. Lung carcinomas in both male and female mice were increased at exposures 40 times those in humans. Combined lung adenomas and carcinomas were increased in male mice at exposures 3 times and in female mice at exposures 40 times those in humans. Tumor development was preceded by pneumocyte proliferation in the lung, which was not observed in rats, dogs, or monkeys administered entecavir, supporting the conclusion that lung tumors in mice may be a species-specific event. Hepatocellular carcinomas were increased in males and combined liver adenomas and carcinomas were also increased at exposures 42 times those in humans. Vascular tumors in female mice (hemangiomas of ovaries and uterus and hemangiosarcomas of spleen) were increased at exposures 40 times those in humans. In rats, hepatocellular adenomas were increased in females at exposures 24 times those in humans; combined adenomas and carcinomas were also increased in females at exposures 24 times those in humans. Brain gliomas were induced in both males and females at exposures 35 and 24 times those in humans. Skin fibromas were induced in females at exposures 4 times those in humans.
Entecavir was clastogenic to human lymphocyte cultures. Entecavir was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains in the presence or absence of metabolic activation, a mammalian-cell gene mutation assay, and a transformation assay with Syrian hamster embryo cells. Entecavir was also negative in an oral micronucleus study and an oral DNA repair study in rats. In reproductive toxicology studies, in which animals were administered entecavir at up to 30 mg/kg for up to 4 weeks, no evidence of impaired fertility was seen in male or female rats at systemic exposures greater than 90 times those achieved in humans at the highest recommended dose of 1 mg/day. In rodent and dog toxicology studies, seminiferous tubular degeneration was observed at exposures 35 times or greater than those achieved in humans. No testicular changes were evident in monkeys.
Microbiology: Mechanism of Action: Entecavir, a guanosine nucleoside analogue with activity against HBV reverse transcriptase (rt), is efficiently phosphorylated to the active triphosphate form, which has an intracellular half-life of 15 hours. By competing with the natural substrate deoxyguanosine triphosphate, entecavir triphosphate functionally inhibits all three activities of the HBV reverse transcriptase: (1) base priming, (2) reverse transcription of the negative strand from the pregenomic messenger RNA, and (3) synthesis of the positive strand of HBV DNA. Entecavir triphosphate is a weak inhibitor of cellular DNA polymerases α, β, and δ and mitochondrial DNA polymerase γ with Ki values ranging from 18 to >160 μM.
Antiviral Activity: Entecavir inhibited HBV DNA synthesis (50% reduction, EC50) at a concentration of 0.004 μM in human HepG2 cells transfected with wild-type HBV. The median EC50 value for entecavir against lamivudine-resistant HBV (rtL180M, rtM204V) was 0.026 μM (range 0.010-0.059 μM).
The coadministration of HIV nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) with entecavir is unlikely to reduce the antiviral efficacy of entecavir against HBV or of any of these agents against HIV. In HBV combination assays in cell culture, abacavir, didanosine, lamivudine, stavudine, tenofovir, or zidovudine were not antagonistic to the anti-HBV activity of entecavir over a wide range of concentrations. In HIV antiviral assays, entecavir was not antagonistic to the cell culture anti-HIV activity of these six NRTIs or emtricitabine at concentrations greater than 100 times the Cmax of entecavir using the 1-mg dose.
Antiviral Activity against HIV: A comprehensive analysis of the inhibitory activity of entecavir against a panel of laboratory and clinical HIV type 1 (HIV-1) isolates using a variety of cells and assay conditions yielded EC50 values ranging from 0.026 to >10 μM; the lower EC50 values were observed when decreased levels of virus were used in the assay. In cell culture, entecavir selected for an M184I substitution in HIV reverse transcriptase at micromolar concentrations, confirming inhibitory pressure at high entecavir concentrations. HIV variants containing the M184V substitution showed loss of susceptibility to entecavir.
Resistance: In Cell Culture: In cell-based assays, 8- to 30-fold reductions in entecavir phenotypic susceptibility were observed for lamivudine-resistant strains. Further reductions (>70-fold) in entecavir phenotypic susceptibility required the presence of amino acid substitutions rtM204I/V with or without rtL180M along with additional substitutions at residues rtT184, rtS202, or rtM250, or a combination of these substitutions with or without an rtI169 substitution in the HBV reverse transcriptase. Lamivudine-resistant strains harboring rtL180M plus rtM204V in combination with the amino acid substitution rtA181C conferred 16- to 122-fold reductions in entecavir phenotypic susceptibility.
In Clinical Studies: Subjects in clinical trials initially treated with entecavir 0.5 mg (nucleoside-naïve, studies AI463022, AI463027, and rollover study AI463901) or 1.0 mg (lamivudine-refractory, studies AI463026, AI463014, AI463015, and rollover study AI463901) and with an on-therapy PCR HBV DNA measurement at or after Week 24 were monitored for resistance.
Nucleoside-naïve subjects: Through Week 240 in nucleoside-naïve studies, genotypic evidence of entecavir resistance-associated (ETVr) substitutions at rtT184, rtS202, or rtM250 was identified in 3 subjects treated with entecavir, 2 of whom experienced virologic breakthrough (see Table 8). These substitutions were observed only in the presence of lamivudine resistance-associated (LVDr) substitutions (rtM204V and rtL180M). (See Table 8.)
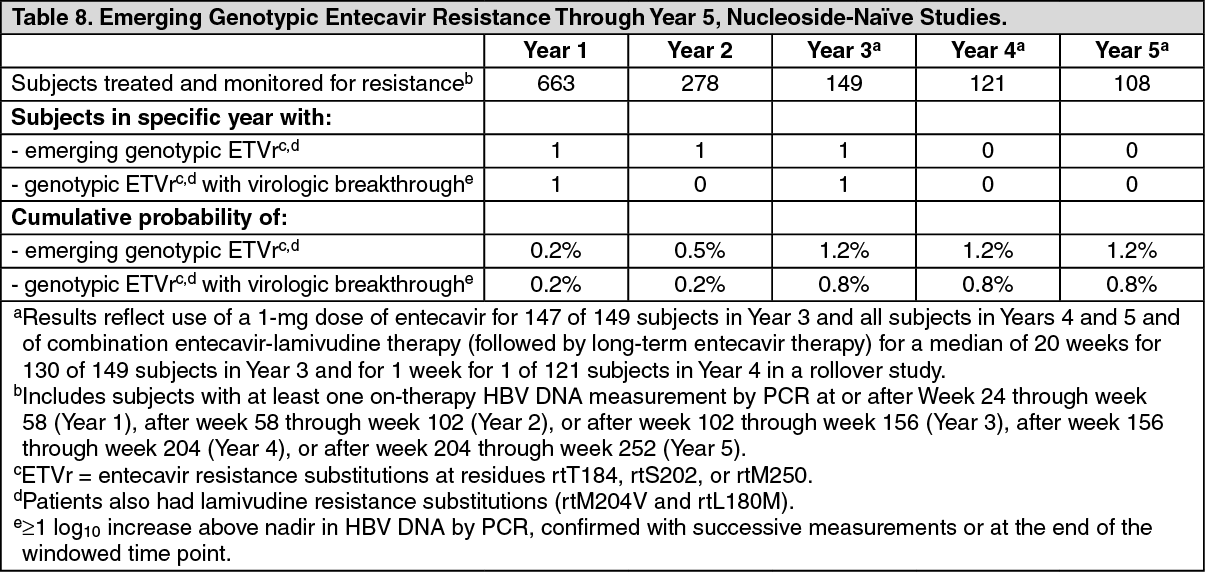 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLamivudine-refractory subjects: ETVr substitutions (in addition to LVDr substitutions rtM204V/I ± rtL180M) were observed at baseline in isolates from 10/187 (5%) lamivudine-refractory subjects treated with entecavir and monitored for resistance, indicating that prior lamivudine treatment can select these resistance substitutions and that they can exist at a low frequency before entecavir treatment. Through Week 240, 3 of the 10 subjects experienced virologic breakthrough (≥1 log10 increase above nadir). Emerging entecavir resistance in lamivudine-refractory studies through Week 240 is summarized in Table 9. (See Table 9.)
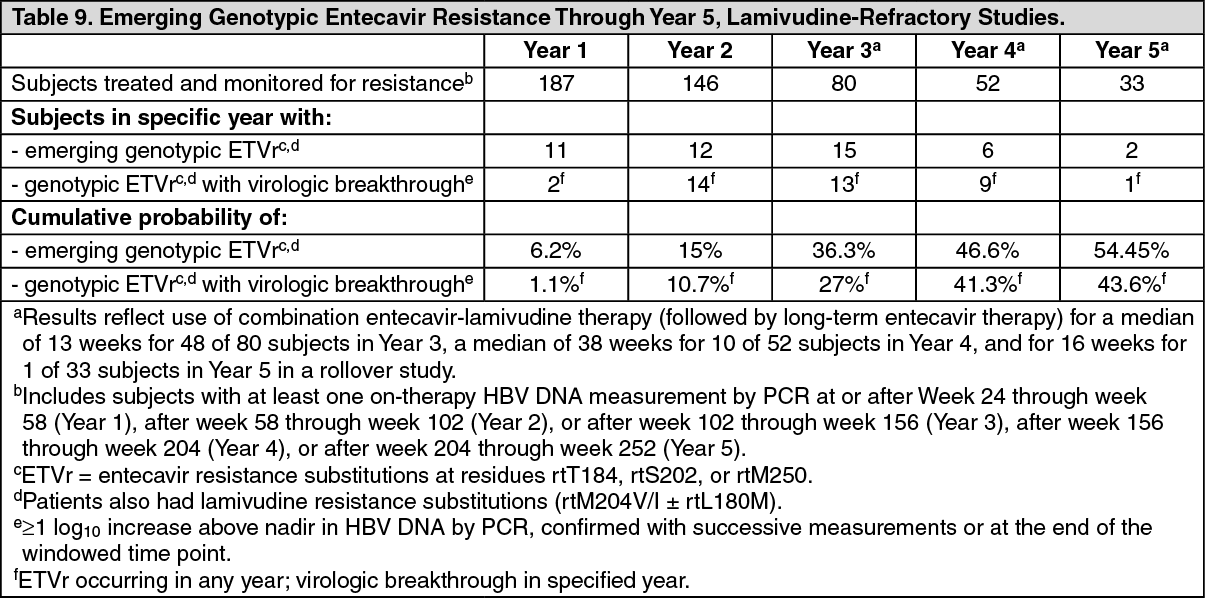 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAmong lamivudine-refractory subjects with baseline HBV DNA <107 log10 copies/mL, 64% (9/14) achieved HBV DNA <300 copies/mL at Week 48. These 14 subjects had a lower rate of genotypic entecavir resistance (cumulative probability 18.8% through 5 years of follow-up) than the overall study population (see Table 9). Also, lamivudine-refractory subjects who achieved HBV DNA <104 log10 copies/mL by PCR at Week 24 had a lower rate of resistance than those who did not (5-year cumulative probability 17.6% [n=50] versus 60.5% [n=135], respectively).
In a post-approval integrated analysis of entecavir resistance data from 17 Phase 2 and 3 clinical trials, an emergent entecavir resistance-associated substitution rtA181C was detected in 5 out of 1461 (0.3%) subjects during treatment with entecavir. This substitution was detected only in the presence of lamivudine resistance-associated substitutions rtL180M plus rtM204V.
Cross-resistance: Cross-resistance has been observed among HBV nucleoside analogues. In cell-based assays, entecavir had 8- to 30-fold less inhibition of HBV DNA synthesis for HBV containing lamivudine and telbivudine resistance substitutions rtM204I/V with or without rtL180M than for wild-type HBV. Substitutions rtM204I/V with or without rtL180M, rtL80I/V, or rtV173L, which are associated with lamivudine and telbivudine resistance, also confer decreased phenotypic susceptibility to entecavir. The efficacy of entecavir against HBV harboring adefovir resistance-associated substitutions has not been established in clinical trials. HBV isolates from lamivudine-refractory subjects failing entecavir therapy were susceptible in cell culture to adefovir but remained resistant to lamivudine. Recombinant HBV genomes encoding adefovir resistance-associated substitutions at either rtN236T or rtA181V had 0.3- and 1.1-fold shifts in susceptibility to entecavir in cell culture, respectively.