


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Mechanism of action: Rimegepant selectively binds with high affinity to the human calcitonin gene-related peptide (CGRP) receptor and antagonises CGRP receptor function.
The relationship between pharmacodynamic activity and the mechanism(s) by which rimegepant exerts its clinical effects is unknown.
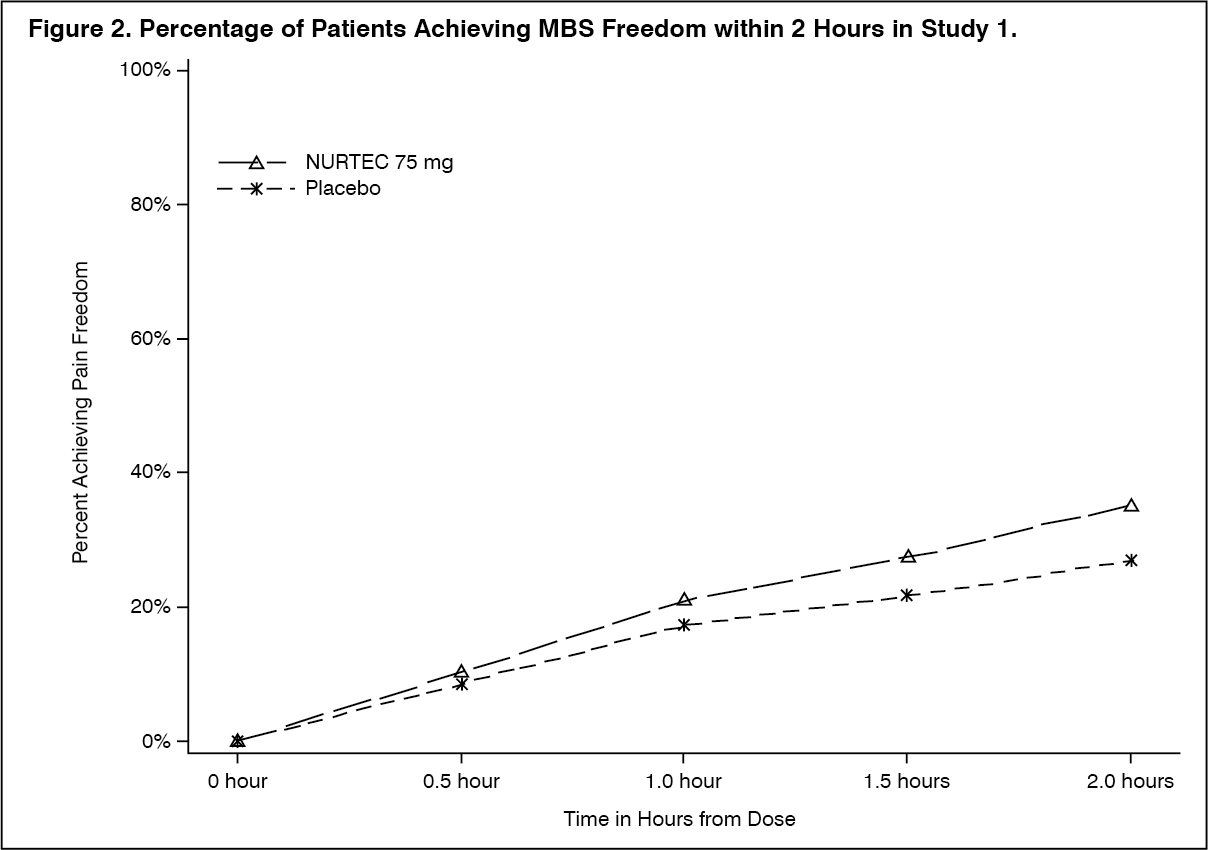
Clinical efficacy: acute treatment: The efficacy of NURTEC for the acute treatment of migraine with and without aura in adults was studied in three randomised, double-blind, placebo-controlled trials (Studies 1-3). Patients were instructed to treat a migraine of moderate to severe headache pain intensity. Rescue medicinal products (i.e., NSAIDs, paracetamol, and/or an antiemetic) was allowed 2 hours after the initial treatment. Other forms of rescue medicinal products such as triptans were not allowed within 48 hours of initial treatment. Approximately 14% of patients were taking preventive medicinal products for migraine at baseline. None of the patients in Study 1 were on concomitant preventive medicinal products that act on the calcitonin gene-related peptide pathway.
The primary efficacy analyses were conducted in patients who treated a migraine with moderate to severe pain. Pain freedom was defined as a reduction of moderate or severe headache pain to no headache pain, and most bothersome symptom (MBS) freedom was defined as the absence of the self-identified MBS (i.e., photophobia, phonophobia, or nausea). Among patients who selected an MBS, the most commonly selected symptom was photophobia (54%), followed by nausea (28%), and phonophobia (15%).
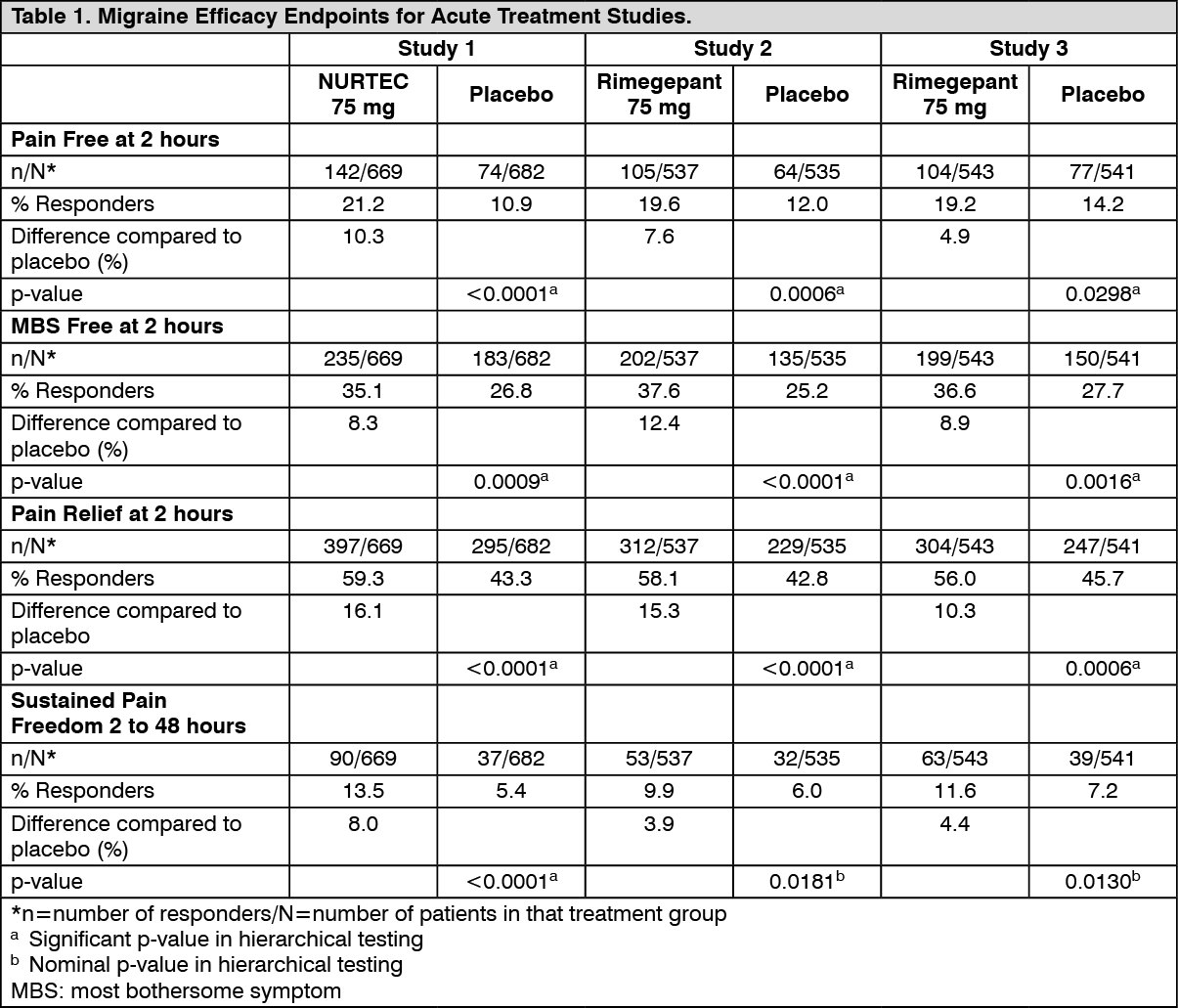
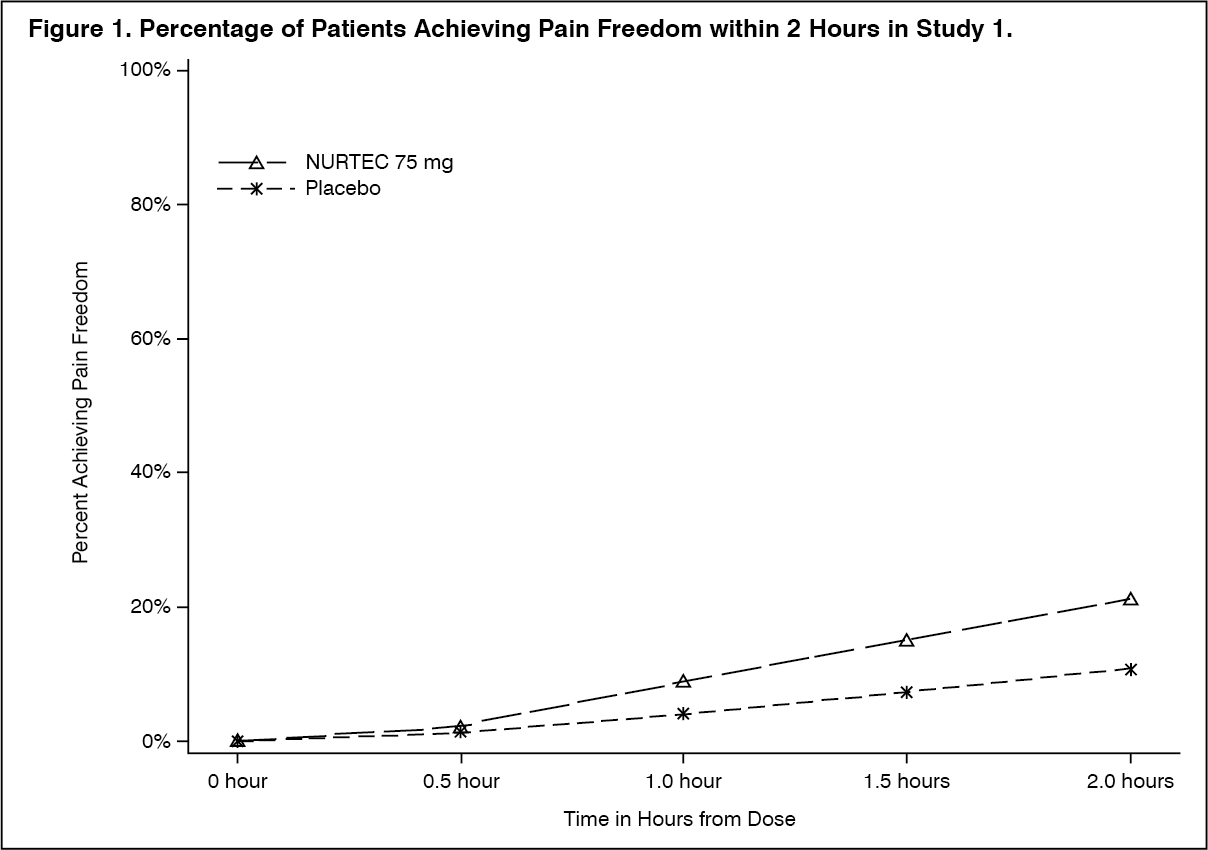
In Study 1, the percentage of patients achieving headache pain freedom and MBS freedom at 2 hours after a single dose was statistically significantly greater in patients who received NURTEC compared to those who received placebo (Table 1). In addition, statistically significant effects of NURTEC compared to placebo were demonstrated for the additional efficacy endpoints of pain relief at 2 hours, sustained pain freedom from 2 to 48 hours, use of rescue medication within 24 hours, and ability to function normally at 2 hours after dosing. Pain relief was defined as a reduction in migraine pain from moderate or severe severity to mild or none. Pivotal single attack, double-blind, placebo-controlled Studies 2 & 3 were conducted in patients with migraine who received one 75 mg rimegepant bioequivalent dosage form. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFigure 1 presents the percentage of patients achieving migraine pain freedom within 2 hours following treatment in Study 1. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFigure 2 presents the percentage of patients achieving MBS freedom within 2 hours in Study 1. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe incidence of photophobia and phonophobia was reduced at 2 hours following administration of NURTEC 75 mg as compared to placebo in all 3 studies.
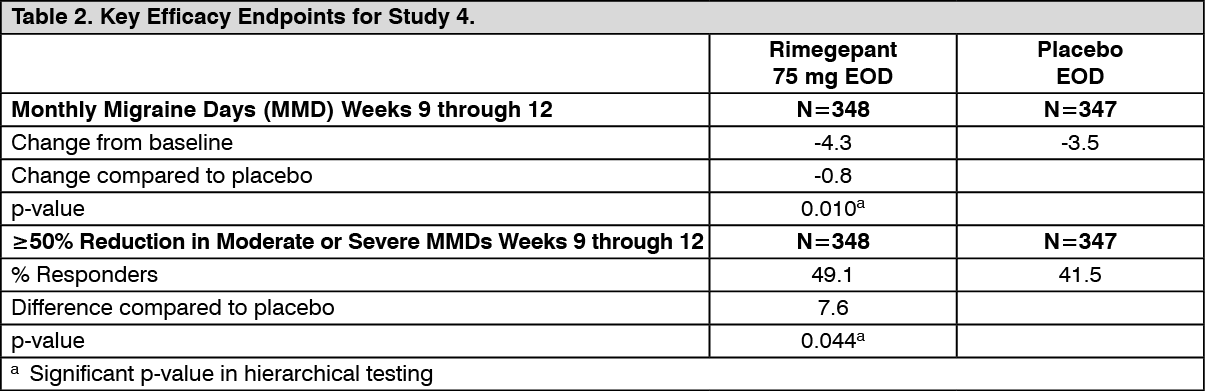
Clinical efficacy: prophylaxis: The efficacy of rimegepant was evaluated as a prophylactic treatment of migraine in a randomised, double-blind, placebo-controlled study (Study 4).
Study 4 included male and female adults with at least a 1-year history of migraine (with or without aura). Patients had a history of 4 to 18 migraine attacks of moderate to severe pain intensity per 4-week period within the 12 weeks prior to the screening visit. Patients experienced an average of 10.9 headache days during the 28-day observational period, which included an average of 10.2 migraine days, prior to randomisation into the study. The study randomised patients to receive rimegepant 75 mg (N=373) or placebo (N=374) for up to 12 weeks. Patients were instructed to take randomised treatment once every other day (EOD) for the 12-week treatment period. Patients were allowed to use other acute treatments for migraine (e.g., triptans, NSAIDs, paracetamol, antiemetics) as needed. Approximately 22% of patients were taking preventive medicinal products for migraine at baseline. Patients were allowed to continue in an open-label extension study for an additional 12 months.
The primary efficacy endpoint for Study 4 was the change from baseline in the mean number of monthly migraine days (MMDs) during Weeks 9 through 12 of the double-blind treatment phase. Secondary endpoints included the achievement of a ≥50% reduction from baseline in monthly moderate or severe migraine days. Rimegepant 75 mg dosed EOD demonstrated statistically significant improvements for key efficacy endpoints compared to placebo, as summarised in Table 2 and shown graphically in Figure 3. (See Table 2 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image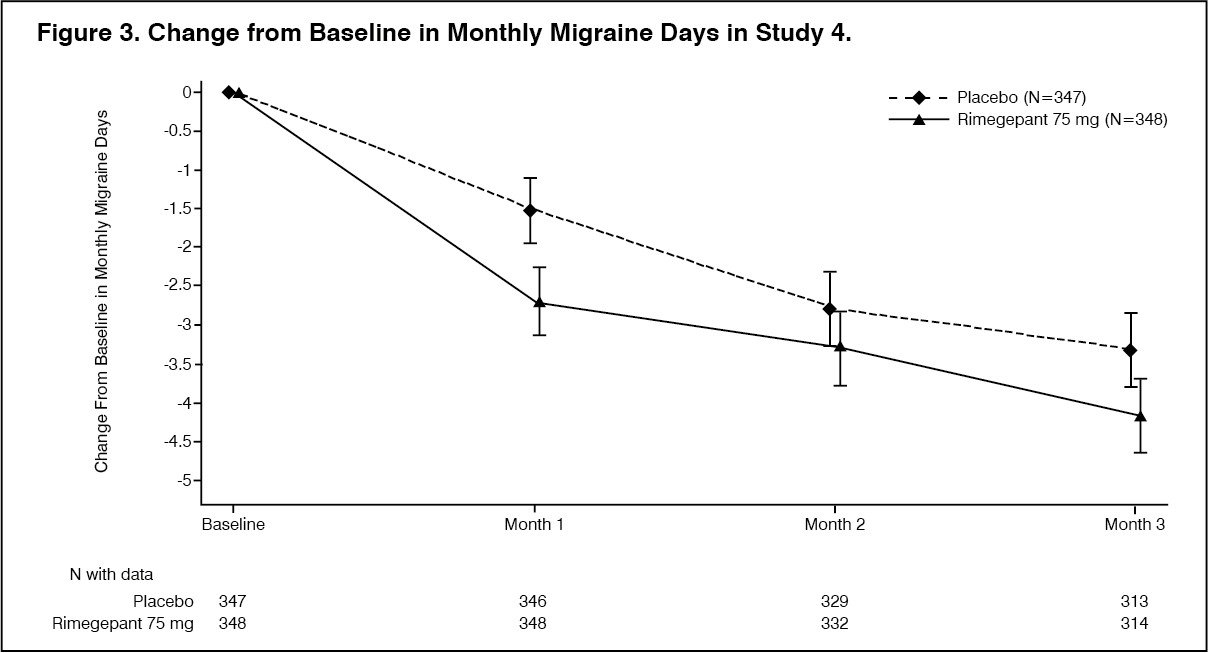 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLong-term efficacy: Patients participating in Study 4 were allowed to continue in an open-label extension study for an additional 12 months. Efficacy was sustained for up to 1 year in an open-label study extension in which patients received rimegepant 75 mg every other day plus as needed on non-scheduled dosing days (Figure 4). A portion composed of 203 patients assigned to rimegepant completed the overall 16-month treatment period. In these patients, the overall mean reduction from baseline in the number of MMDs averaged over the 16-month treatment period was 6.2 days. (See Figure 4.)
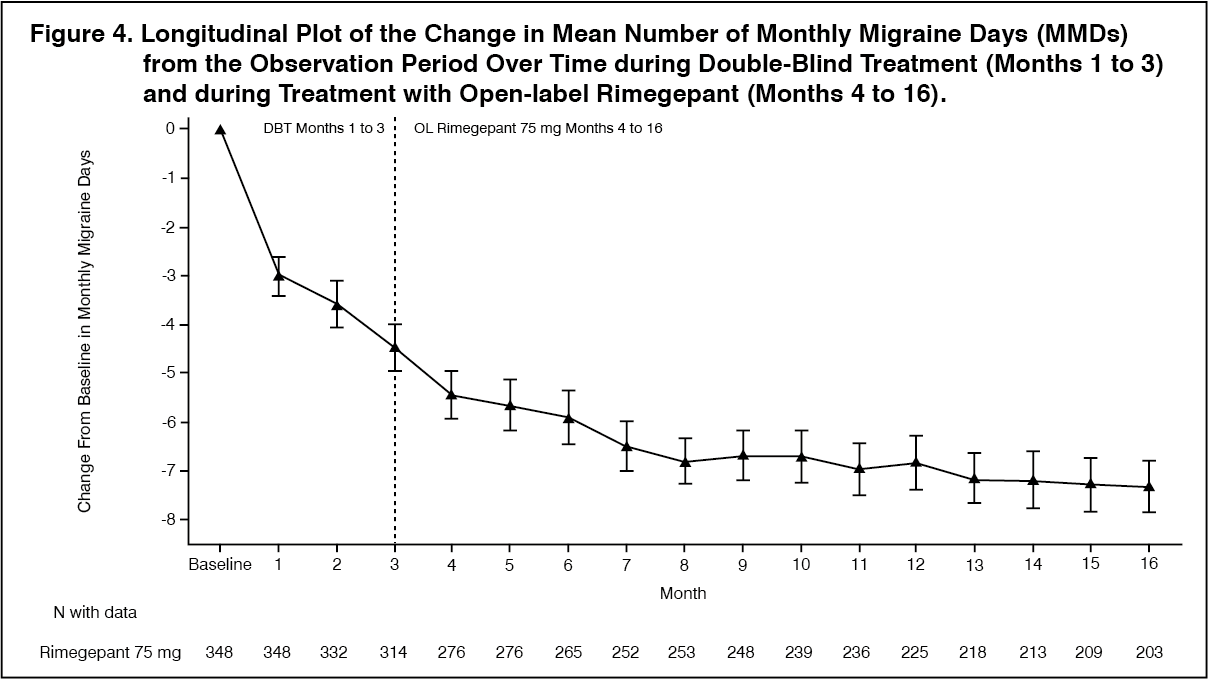 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Following oral administration, rimegepant is absorbed with the maximum concentration at 1.5 hours. Following a supratherapeutic dose of 300 mg, the absolute oral bioavailability of rimegepant was approximately 64%.
Effects of food: Following administration of rimegepant under fed conditions with a high-fat or low-fat meal, Tmax was delayed by 1 to 1.5 hours. A high-fat meal reduced Cmax by 42 to 53% and AUC by 32 to 38%. A low-fat meal reduced Cmax by 36% and AUC by 28%. Rimegepant was administered without regard to food in clinical safety and efficacy studies.
Distribution: The steady state volume of distribution of rimegepant is 120 L. Plasma protein binding of rimegepant is approximately 96%.
Biotransformation: Rimegepant is primarily metabolised by CYP3A4 and to a lesser extent by CYP2C9. Rimegepant is primarily eliminated in unchanged form (~77% of the dose) with no major metabolites (i.e., >10%) detected in plasma.
Based on in vitro studies, rimegepant is not an inhibitor of CYP1A2, 2B6, 2C9, 2C19, 2D6, or UGT1A1 at clinically relevant concentrations. However, rimegepant is a weak inhibitor of CYP3A4 with time-dependent inhibition. Rimegepant is not an inducer of CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
Elimination: The elimination half-life of rimegepant is approximately 11 hours in healthy subjects. Following oral administration of [14C]-rimegepant to healthy male subjects, 78% of the total radioactivity was recovered in faeces and 24% in urine. Unchanged rimegepant is the major single component in excreted faeces (42%) and urine (51%).
Transporters: In vitro, rimegepant is a substrate of P-gp and BCRP efflux transporters. Inhibitors of P-gp and BCRP efflux transporters may increase plasma concentrations of rimegepant (see Interactions).
Rimegepant is not a substrate of OATP1B1 or OATP1B3. Considering its low renal clearance, rimegepant was not evaluated as a substrate of the OAT1, OAT3, OCT2, MATE1, or MATE2-K.
Rimegepant is not an inhibitor of P-gp, BCRP, OAT1, or MATE2-K at clinically relevant concentrations. It is a weak inhibitor of OATP1B1 and OAT3.
Rimegepant is an inhibitor of OATP1B3, OCT2, and MATE1. Concomitant administration of rimegepant with metformin, a MATE1 transporter substrate, resulted in no clinically significant impact on either metformin pharmacokinetics or on glucose utilisation. No clinical drug interactions are expected for rimegepant with OATP1B3 or OCT2, at clinically relevant concentrations.
Linearity/non-linearity: Rimegepant exhibits greater than dose proportional increases in exposure following single oral administration, which appears to be related to a dose-dependent increase in bioavailability.
Age, sex, weight, race, ethnicity: No clinically significant differences in the pharmacokinetics of rimegepant were observed based on age, sex, race/ethnicity, body weight, migraine status, or CYP2C9 genotype.
Renal impairment: In a dedicated clinical study comparing the pharmacokinetics of rimegepant in subjects with mild (estimated creatinine clearance [CLcr] 60-89 mL/min), moderate (CLcr 30-59 mL/min), and severe (CLcr 15-29 mL/min) renal impairment to that of normal subjects (healthy pooled control), a less than 50% increase in total rimegepant exposure was observed following a single 75 mg dose. The unbound AUC of rimegepant was 2.57-fold higher in subjects with severe renal impairment. NURTEC has not been studied in patients with end-stage renal disease (CLcr <15 mL/min).
Hepatic impairment: In a dedicated clinical study comparing the pharmacokinetics of rimegepant in subjects with mild, moderate, and severe hepatic impairment to that of normal subjects (healthy matched control), the exposure of rimegepant (unbound AUC) following a single 75 mg dose was 3.89-fold higher in subjects with severe impairment (Child-Pugh class C). There were no clinically meaningful differences in the exposure of rimegepant in subjects with mild (Child-Pugh class A) and moderate hepatic impairment (Child-Pugh class B) compared to subjects with normal hepatic function.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for rimegepant in humans based on conventional studies of safety pharmacology, repeat-dose toxicity, genotoxicity, phototoxicity, reproduction or development, or carcinogenic potential.
Rimegepant-related effects at higher doses in repeat-dose studies included hepatic lipidosis in mice and rats, intravascular haemolysis in rats and monkeys, and emesis in monkeys. These findings were observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use (≥12 times [mice] and ≥49 times [rats] for hepatic lipidosis, ≥95 times [rats] and ≥9 times [monkeys] for intravascular haemolysis, and ≥37 times for emesis [monkeys]).
Oral administration of rimegepant to male and female rats prior to and during mating, and continuing in females to gestation day 7, resulted in reduced fertility at the highest dose (150 mg/kg/day) tested.
The no-effect dose for impairment of fertility and early embryonic development in rats of 60 mg/kg/day was associated with plasma drug exposures (AUC) approximately 30 times that in humans at the MRHD of 75 mg/day.